Radiologia Brasileira - Publicação Científica Oficial do Colégio Brasileiro de Radiologia
AMB - Associação Médica Brasileira CNA - Comissão Nacional de Acreditação
 Vol. 47 nº 3 - Maio / Jun. of 2014
Vol. 47 nº 3 - Maio / Jun. of 2014
|
RELATO DE CASO
|
|
|
|
|
Autho(rs): Junio Marcos Fonseca1; Luciana Mendes Araújo Borém2 |
|
|
Descritores: Síndrome; Malformação; Tomografia computadorizada. |
|
|
Resumo: INTRODUÇÃO
A síndrome do "crânio em folha de trevo" é uma configuração anormal da calvária classificada como uma craniossinostose, em que ocorre ossificação prematura das suturas cranianas. Trata-se de deformidade caracterizada por acentuado alargamento da cabeça, com configuração trilobulada da visão frontal, lembrando um trevo de três folhas(1). Devese a uma alteração severa do desenvolvimento do crânio, com sinostose prematura de algumas suturas cranianas(1) - mais comumente a coronal e a lambdóide(2) - associada a hidrocefalia, resultando em acentuado abaulamento superior da cabeça na região da fontanela anterior e lateralmente nos polos temporais, configurando o aspecto típico em "folha de trevo"(1). Tem sido reportada nas formas sindrômica e não sindrômica. Por apresentar anomalias tanto na calvária quanto na base do crânio e na face, trata-se de uma das craniossinostoses de tratamento mais complexo atualmente(1). O primeiro relato na literatura se deu em 1973, e desde então apenas algumas dezenas de casos foram documentados no mundo(3). O presente caso descreve uma doença craniofacial grave, conhecida como "crânio em folha de trevo". RELATO DO CASO Paciente do sexo feminino, nascida de parto cesariano no dia 4/1/2011, pesando 3.815 g e com Apgar 7 e 8 no primeiro e quinto minutos de vida, respectivamente. Ao exame físico notou-se a presença de múltiplas malformações, com alteração da configuração craniofacial, implantação baixa das orelhas, hipertelorismo, exoftalmia, onfalocele e polidactilia (Figura 1). Foram realizadas radiografia de tórax, que mostrou dextrocardia (Figura 2), e tomografia computadorizada, que revelou alteração da configuração da calota craniana, com aspecto lobulado típico em "folha de trevo", abaulamento das fossas cranianas médias e dilatação do sistema ventricular. Sinais de fechamento das suturas sagitais, coronais e lambdoides também foram observados (Figura 3). A paciente apresentou paradas cardiorrespiratórias, evoluindo para o óbito no dia 7/1/2011. 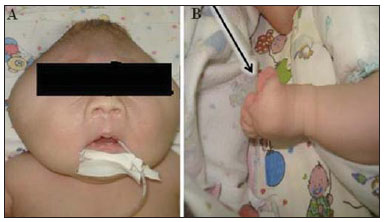 Figura 1. A: Demonstração das alterações craniofaciais típicas. B: Sindactilia (seta). 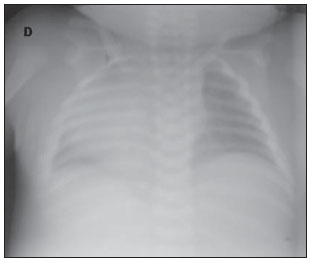 Figura 2. Radiografia do tórax mostrando dextrocardia. 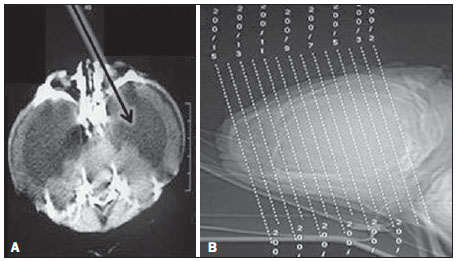 Figura 3. A: Aspecto trilobulado do crânio, com hidrocefalia e abaulamento das fossas médias (seta). B: Scout do crânio demonstrando o acentuado crescimento superior da cabeça na região da fontanela anterior. DISCUSSÃO A síndrome do crânio em formato de "folha de trevo" é uma forma rara de craniossinostose com características clínicas que consistem em crânio trilobulado, exoftalmia, implantação baixa das orelhas e obstrução das vias aéreas superiores. Hidrocefalia é também comum, embora seu mecanismo patogênico seja multifatorial(4). Trata-se de uma anomalia congênita que pode estar presente como um defeito isolado, mas que geralmente classifica-se em síndromes disostóticas como acondroplasia, disostose craniofacial de Crouzon, síndromes de Apert ou de Pfeiffer(2). Até 1981, apenas 30 casos haviam sido publicados na literatura mundial, sendo o primeiro em 1973 na literatura oftálmica(3). A etiopatogenia exata da síndrome não é muito clara(3), com teorias que envolvem ossificação óssea membranosa e/ou endocondral alterada, processo condrodisplásico generalizado(2) e uma possível origem vascular relacionada a reabsorção osteoclástica anormal(5). Recentemente, investigações genéticas têm contribuído para avançar na compreensão da base molecular de algumas síndromes de craniossinostose, destacando-se mutações nos genes FGFR1, FGFR2, FGFR3, TWIST e MSX2(6). Seu diagnóstico pode ser realizado no período pré-natal por ultrassonografia, que detecta a morfologia alterada do crânio e a hidrocefalia. Tradicionalmente, o diagnóstico ocorre no segundo trimestre, em exames de rotina. No entanto, com o crescente emprego da ultrassonografia obstétrica no primeiro trimestre, essas alterações podem ser detectadas cada vez mais precocemente na gestação(7). Após o nascimento, quando uma configuração anormal da calvária é detectada, a avaliação radiológica é necessária para caracterizar a deformidade e guiar o procedimento cirúrgico corretivo. Há melhora significativa no prognóstico das crianças afetadas quando o diagnóstico e a intervenção cirúrgica ocorrem o mais cedo possível. A tomografia computadorizada com reconstruções tridimensionais contribui para a avaliação das deformidades ósseas craniofaciais e das alterações intracranianas associadas, demonstrando ser ferramenta valiosa para o prognóstico e planejamento cirúrgico do paciente(8). O caso descrito no presente artigo não foi classificado em nenhuma síndrome específica, e a instabilidade cardiorrespiratória da paciente impediu tentativas de intervenção cirúrgica. Seu óbito precoce ilustra como a associação de malformações pode piorar o prognóstico dessa condição. Apesar do desfecho desfavorável de muitos casos, avanços nas técnicas cirúrgica, anestésica e de terapia intensiva tornaram esta condição, antes encarada como não tratável, em potencialmente tratável, com resultados estéticos e neurológicos aceitáveis(2). Destaca-se a importância do diagnóstico e da caracterização por imagem dessa síndrome, sobretudo no período prénatal, para que o tratamento seja planejado e instituído o mais precocemente possível. REFERÊNCIAS 1. Iannaccone G, Gerlini G. The so-called "cloverleaf skull syndrome". A report of three cases with a discussion of its relationships with thanatophoric dwarfism and the craniostenoses. Pediat Radiol. 1974;2:175-84. 2. Manjila S, Chim H, Eisele S, et al. History of the Kleeblattschädel deformity: origin of concepts and evolution of management in the past 50 years. Neurosurg Focus. 2010;29:E7. 3. Ghose S, Mehta U. The Kleeblattschadel (cloverleaf skull) syndrome. Indian J Ophthalmol. 1986;34:61-6. 4. Ito S, Matsui K, Ohsaki E, et al. A cloverleaf skull syndrome probably of Beare-Stevenson type associated with Chiari malformation. Brain Dev. 1996;18:307-11. 5. Dambrain R, Freund M, Verellen G, et al. Considerations about the cloverleaf skull. J Craniofac Genet Dev Biol. 1987;7:387-401. 6. Arduino-Meirelles AP, Lacerda CBF, Gil-da-Silva-Lopes VL. Aspectos sobre desenvolvimento de linguagem oral em craniossinostoses sindrômicas. Pró-Fono. 2006;18:213-20. 7. Chitty LS, Khalil A, Barrett AN, et al. Safe, accurate, prenatal diagnosis of thanatophoric dysplasia using ultrasound and free fetal DNA. Prenatal Diagn. 2013;33:416-23. 8. Trad CS, Rosique RG. Craniossinostoses primárias: ensaio iconográfico. Radiol Bras. 2005;38:377-80. 1. Pós-graduado em Biomedicina, Coordenador Técnico do Setor de Imaginologia da Fundação Dilson Godinho, Montes Claros, MG, Brasil 2. Mestre, Doutoranda em Ciências da Saúde, Médica Radiologista do Hospital Santa Casa de Montes Claros, Montes Claros, MG, Brasil Endereço para correspondência: Junio Marcos Fonseca Rua Guiana Francesa, 200, Bairro João Alves Montes Claros, MG, Brasil, 39402-307 E-mail: juniofonseca@hotmail.com Recebido para publicação em 28/1/2013. Aceito, após revisão, em 12/8/2013. Trabalho realizado no Hospital Santa Casa de Montes Claros, Montes Claros, MG, Brasil. |
|
Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554