Radiologia Brasileira - Publicação Científica Oficial do Colégio Brasileiro de Radiologia
AMB - Associação Médica Brasileira CNA - Comissão Nacional de Acreditação
 Vol. 45 nº 4 - Jul. / Ago. of 2012
Vol. 45 nº 4 - Jul. / Ago. of 2012
|
RELATO DE CASO
|
|
|
|
|
Autho(rs): Josie Naomi Iyeyasu1; Ana Carolina Marcos Vaz2; Fabiano Reis3; João Altemani4; Luciano de Souza Queiroz5; Keila Monteiro de Carvalho6 |
|
|
Descritores: Histiocitose; Células de Langerhans; Órbita. |
|
|
Resumo: INTRODUÇÃO
A histiocitose de células de Langerhans é uma doença rara, que normalmente afeta crianças e pode acometer diversas partes do corpo. O seu diagnóstico é feito com base nos exames histológico e imuno-histoquímico das lesões. O tratamento depende da forma de apresentação da doença. O objetivo do presente estudo é descrever o caso de um paciente de 63 anos de idade acometido pela doença, bem como apresentar seus exames de imagens e a evolução do caso. Ao final, é apresentado breve resumo da literatura sobre a doença. RELATO DO CASO Paciente do sexo masculino, de 63 anos, com queixa de proptose do olho direito há seis meses. Antecedentes pessoais: tabagista e etilista. A triagem para doenças infectocontagiosas foi negativa. O exame físico, à exceção da proptose à direita, não apresentou alterações. Foi realizada tomografia computadorizada de crânio (Figura 1), que revelou uma lesão comprometendo a parede lateral da órbita (com destruição óssea) e região intraorbitária direita, estendendo-se externamente para a região perizigomática. Observou-se proptose do globo ocular direito e invasão da musculatura extrínseca lateral da órbita desse lado. 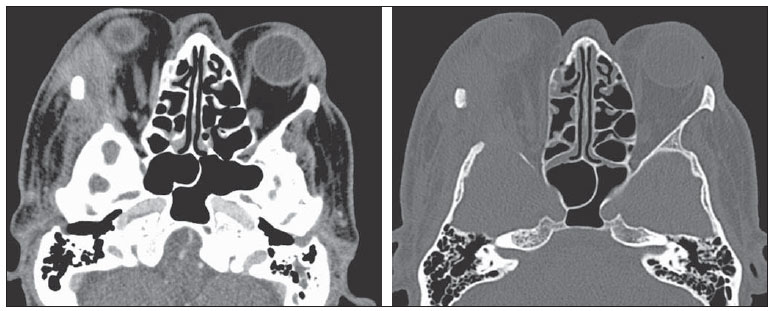 Figura 1. Imagens axiais de tomografia computadorizada de crânio com contraste do paciente. Lesão expansiva intraorbitária com extensão para os espaços intra e extraconais, tecidos moles extracranianos e erosão óssea da parede lateral (osso zigomático e asa maior do esfenoide) da órbita direita. O paciente foi, então, submetido a exérese completa da lesão. O exame histopatológico revelou proliferação de padrão histiocítico macrofágico abundante, com células epitelioides e gigantócitos multinucleados e algumas figuras de mitose, compatível com histiocitose de células de Langerhans (Figura 2). 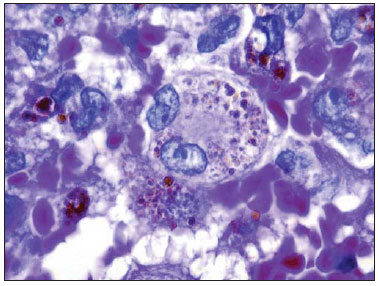 Figura 2. Coloração hematoxilina-eosina demonstra células de Langerhans fagocitando hemossiderina. Elas são reconhecíveis pelo núcleo irregular com contorno arredondado ou alongado, frequentemente com clivagens e endentações, cromatina frouxa e bem distribuída. O exame imuno-histoquímico apresentou-se positivo para CD1a, CD 68, HAM56 e S-100 nas células neoplásicas, negativo para HMB45, AE1, AE3, CD20, CD3 e positivo para KI67 em 5-10% dos núcleos, corroborando a hipótese diagnóstica de histiocitose de células de Langerhans (Figura 3). 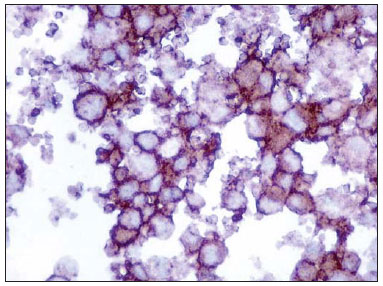 Figura 3. Estudo imuno-histoquímico demonstra positividade para CD1a em padrão membrana, condição sine qua non para diagnóstico da histiocitose de células de Langerhans. O paciente foi, então, submetido à pesquisa de outros focos de acometimento pela histiocitose, que revelou lesão do quinto arco costal esquerdo e comprometimento pulmonar característico da histiocitose (Figura 4). O paciente realizou acompanhamento com equipe de hematologia, tendo sido submetido a dois ciclos de quimioterapia com vimblastina, etoposide e prednisona. Evoluiu a óbito em fevereiro de 2012, em decorrência de complicações advindas de sua doença pulmonar. 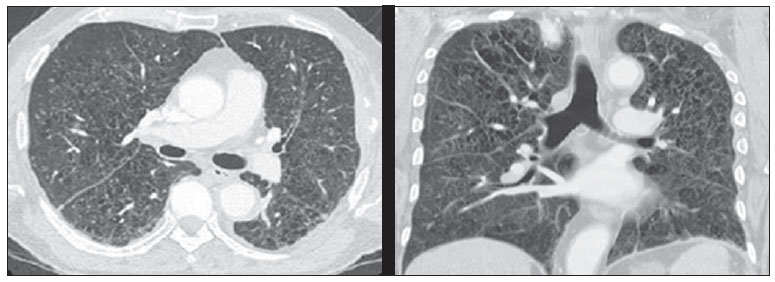 Figura 4. Tomografia computadorizada de tórax do paciente evidenciando múltiplos cistos difusamente distribuídos pelo parênquima pulmonar, com paredes irregulares e dimensões variáveis ("cistos bizarros"), associados a micronódulos difusos, aspecto característico do acometimento pulmonar pela histiocitose de células de Langerhans. DISCUSSÃO A histiocitose de células de Langerhans é uma doença rara (incidência de dois a cinco casos em um milhão)(1), caracterizada pela proliferação de células de Langerhans(2), que acomete mais comumente crianças de um a três anos de idade do sexo masculino(1). A etiologia da doença continua incerta. Acredita-se que a doença seja consequência de uma desregulação imunológica, resultando na produção excessiva de citocinas e prostaglandinas, que ocasionam danos em diversos órgãos(1). A doença pode se manifestar localmente ou acometer múltiplos órgãos. O quadro clínico vai depender do grau de disfunção dos órgãos atingidos(1). As principais complicações da doença são a disfunção hipofisária, com destaque para o diabetes insipidus(3), e as doenças neurodegenerativas(4). A manifestação oftalmológica mais comum é a presença de lesão óssea isolada da órbita(5), que é considerada, assim como a presença de lesões no osso mastoide e no osso temporal(6), fator de risco para o surgimento de complicações no sistema nervoso central(7). A histiocitose de células de Langerhans, principalmente na forma disseminada, pode estar associada a outras condições, por exemplo, a leucemia e o linfoma de Hodgkin e não Hodgkin, história de infecção neonatal, exposição a solventes, doenças da tireoide, doenças infecciosas (adenovírus, parvovírus, Epstein-Barr, citomegalovírus, HIV e HTLV) e tabagismo(1), como pode ter ocorrido em nosso caso. O exame de imagem de escolha para avaliação do sistema nervoso central é a ressonância magnética, por ser o mais sensível na detecção de lesões, embora não haja um padrão radiológico característico da doença(3). O diagnóstico da histiocitose de células de Langerhans pode ser feito pelo exame histológico das lesões (diagnóstico presuntivo) ou pelo estudo imuno-histoquímico. À microscopia óptica, é observado um infiltrado monoclonal neoplásico de células de Langerhans, com núcleo lobulado de cromatina fina e citoplasma eosinofílico moderadamente abundante, com algumas atipias nucleares e atividade mitótica variável(3). Já o exame imuno-histoquímico revela a presença de grânulos de Birbeck, positividade para S-100 e CD1a (sendo este o diagnóstico de certeza)(1), como ocorreu no caso aqui relatado. O tratamento da histiocitose de células de Langerhans é a intervenção local(6) em casos em que há apenas uma única lesão, ficando o tratamento sistêmico reservado para os casos em que há resposta incompleta, reativação da lesão, ou surgimento de outras lesões(7), sendo a quimioterapia com vimblastina o padrão ouro(2), como ocorreu em nosso caso. O prognóstico depende do grau de acometimento. Pacientes com doença local normalmente apresentam bom prognóstico, enquanto os com doença disseminada e os que têm acometimento do sistema nervoso central apresentam pior prognóstico(6). Podemos concluir que a histiocitose de células de Langerhans é uma doença rara e grave, podendo acometer diversos órgãos. Portanto, ela deve entrar na lista de diagnósticos diferenciais de pacientes com proptose, já que o acometimento orbitário indica provável comprometimento do sistema nervoso central, ou seja, pior prognóstico. Consideramos o caso descrito importante por ser bem ilustrativo em relação à evolução desfavorável do paciente, mesmo com tratamento adequado, servindo para enfatizar a importância do diagnóstico precoce da doença. Além disso, trata-se de um caso raro, já que o paciente foi acometido pela doença em idade avançada, quando o mais freqüente é o acometimento na infância, além de sua lesão orbital ser um local raramente atingido pela doença. REFERÊNCIAS 1. Duda-Szymańska J, Wierzchniewska-ºawska A. Langerhans cell histiocytosis in a 3-year-old girl: a case report and literature review. Pol J Pathol. 2009;60:134-7. 2. Ng Wing Tin S, Martin-Duverneuil N, Idbaih A, et al. Efficacy of vinblastine in central nervous system Langerhans cell histiocytosis: a nationwide retrospective study. Orphanet J Rare Dis. 2011;6:83. 3. Grois N, Tsunematsu Y, Barkovich AJ, et al. Central nervous system disease in Langerhans cell histiocytosis. Br J Cancer Suppl. 1994;23:S24-S28. 4. Imashuku S. High dose immunoglobulin (IVIG) may reduce the incidence of Langerhans cell histiocytosis (LCH)-associated central nervous system involvement. CNS Neurol Disord Drug Targets. 2009;8:380-6. 5. Margo CE, Goldman DR. Langerhans cell histiocytosis. Surv Ophthalmol. 2008;53:332-58. 6. Allen CE, McClain KL. Langerhans cell histiocytosis: a review of past, current and future therapies. Drugs Today (Barc). 2007;43:627-43. 7. Harris GJ. Langerhans cell histiocytosis of the orbit: a need for interdisciplinary dialogue. Am J Ophthalmol. 2006;141:374-8. 1. Mestre, Médica Oftalmologista do Hospital de Clínicas da Universidade Estadual de Campinas (HC-Unicamp), Campinas, SP, Brasil. 2. Médica Residente do Departamento de Radiologia da Faculdade de Ciências Médicas da Universidade Estadual de Campinas (FCM-Unicamp), Campinas, SP, Brasil. 3. Doutor, Docente do Departamento de Radiologia da Faculdade de Ciências Médicas da Universidade Estadual de Campinas (FCM-Unicamp), Campinas, SP, Brasil. 4. Doutor, Médico Assistente do Departamento de Radiologia da Faculdade de Ciências Médicas da Universidade Estadual de Campinas (FCM-Unicamp), Campinas, SP, Brasil. 5. Doutor, Docente do Departamento der Anatomia Patológica da Faculdade de Ciências Médicas da Universidade Estadual de Campinas (FCM-Unicamp), Campinas, SP, Brasil. 6. Livre-docente, Docente do Departamento de Oftalmo-Otorrino da Faculdade de Ciências Médicas da Universidade Estadual de Campinas (FCM-Unicamp), Campinas, SP, Brasil. Endereço para correspondência: Dr. Fabiano Reis Hospital de Clínicas da Universidade Estadual de Campinas (HC-Unicamp) Rua Vital Brasil, 251, Cidade Universitária Zeferino Vaz Campinas, SP, Brasil, 13083-888 Caixa Postal 6142 E-mail: fabianoreis2@gmail.com Recebido para publicação em 21/3/2012. Aceito, após revisão, em 15/6/2012. Trabalho realizado no Hospital de Clínicas da Universidade Estadual de Campinas (HC-Unicamp), Campinas, SP, Brasil. |
|
Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554