Radiologia Brasileira - Publicação Científica Oficial do Colégio Brasileiro de Radiologia
AMB - Associação Médica Brasileira CNA - Comissão Nacional de Acreditação
 Vol. 40 nº 1 - Jan. / Fev. of 2007
Vol. 40 nº 1 - Jan. / Fev. of 2007
|
RELATO DE CASO
|
|
|
|
|
Autho(rs): Sizenildo da Silva Figueirêdo, Jobelino Sousa de Araújo, José Eduardo Marini Kozan, Nilton César Lima dos Santos, Valessa Tanganeli |
|
|
Descritores: Condrodisplasia punctata, Displasia óssea, Desordens peroxissomais |
|
|
Resumo:
INTRODUÇÃO Epífises puntiformes são um sinal radiológico presente em várias displasias ósseas, bem como associadas a diferentes desordens metabólicas, teratogênicas e cromossômicas. Estas calcificações puntiformes podem resultar em retardo do processo de ossificação endocondral, crescimento reduzido e deformidade dos ossos afetados(1). Dentre as causas de epífises puntiformes destacamos a condrodisplasia punctata (CDP), cujas variantes fenotípicas estão relacionadas e são determinadas pelo tipo de transmissão genética. A CDP corresponde a um grupo heterogêneo de displasias caracterizado por calcificações puntiformes da cartilagem (principalmente epifisárias), freqüentemente associadas a encurtamento dos membros, catarata, ictiose, alopécia, alterações do sistema nervoso, deficiência mental e do crescimento(2–5). Há altos índices de natimortalidade ou de mortalidade durante o primeiro ano de vida, em conseqüência de anomalias associadas ou de doenças intercorrentes(3). Atualmente, o diagnóstico da CDP é realizado por meio de análise clínica, concomitante a achados bioquímicos e radiológicos. Embora apresente características clínicas e radiológicas bastante evidentes que podem determinar um diagnóstico preciso, outras doenças devem ser consideradas devido à semelhança de seus achados. A família da CDP inclui a forma autossômica dominante (doença de Conradi-Hünnermann), a forma autossômica recessiva (rizomélica), as formas ligadas ao X recessivo (CDPX1) e dominante (CDPX2 ou Conradi-Hünnermann-Happle)(5) . As formas autossômicas resultam de distúrbio do metabolismo peroxissomal(4,6), a forma dominante ligada ao X (CDPX2) é causada por defeito na via da biossíntese do colesterol e a forma recessiva ligada ao X (CDPX1) resulta de defeitos no gene E da arilsulfatase(4). Os peroxissomas são organelas presentes no citoplasma de várias células tissulares (prevalentemente no fígado, rins e fibroblastos) e contêm complexo sistema enzimático com funções diversas, tais como: oxidação de vários compostos e produção de H2O2; biossíntese de ácido biliar, colesterol e plasmalogênio celular; b-oxidação dos ácidos graxos de cadeia longa; metabolismo de lisina e síntese/metabolismo do gliossilato(6). Apresentamos um caso de CDP, descrevendo as duas principais variantes (autossômicas recessiva e dominante), com base em breve revisão da literatura vigente sobre etiopatogenia, manifestações clínico-radiológicas e laboratoriais.
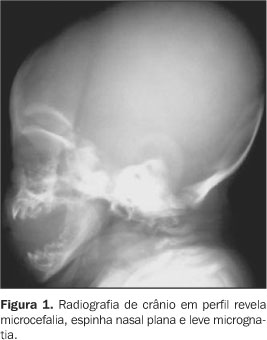
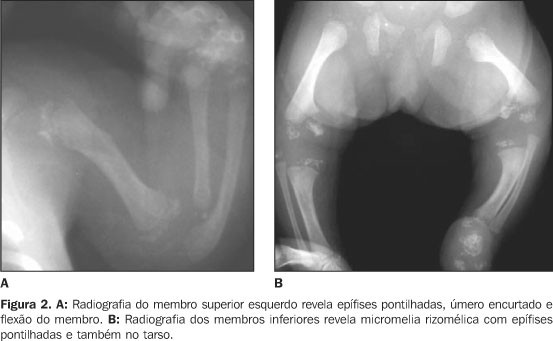
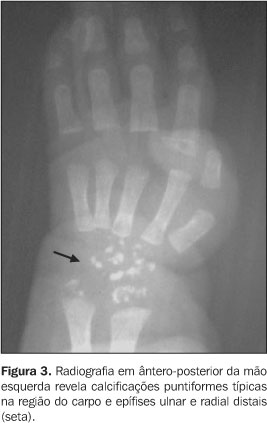
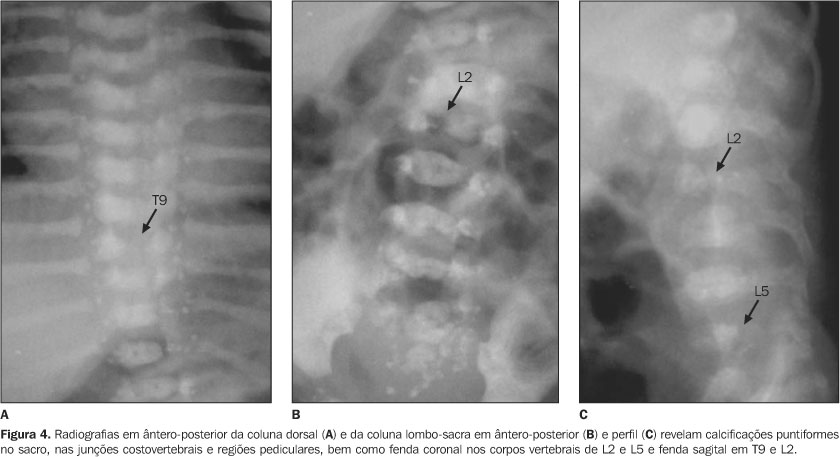
RELATO DO CASO Infante do sexo feminino, dois meses de idade, foi encaminhada ao Serviço de Pediatria do Hospital de Base Ary Pinheiro, Porto Velho, RO, por causa de sua respiração ofegante (taquipnéia) e história de engasgos freqüentes após mamadas, bem como evidentes alterações anatômicas em face e membros. Foi mantida em incubadora termorregulada e O2 circulante, onde ficou internada durante 11 dias. Filha de pais não consanguíneos (mãe primípara) e nascida pré-termo, apresentou uma série de achados clínicos e radiológicos que sugeriram o diagnóstico de CDP. A mãe negou abuso de drogas ou álcool ou exposição ao warfarim (causas teratogênicas conhecidas de epífises puntiformes). Clinicamente, apresentava alterações cutâneas como pele frouxa e escamosa (ictiose), focos irregulares de alopécia, encurtamento de membros em flexão permanente e espasticidade presentes, micrognatia, base nasal achatada, tórax atarracado, bossa serossanguínea, além da já mencionada taquipnéia e gemência. Ausculta cardiopulmonar revelou discretas crepitações bibasais e abdome sem alterações. À somatometria apresentou peso de 1.755 g, 37 cm de estatura, perímetro cefálico e perímetro torácico ambos de 25 cm (abaixo do quinto percentil). Exames laboratoriais de rotina e para pesquisa de infecções congênitas estavam normais. Ao estudo radiográfico observamos: a) micrognatia; b) espinha nasal achatada (Figura 1); c) encurtamento proximal simétrico e bilateral dos ossos dos membros inferiores e superiores (padrão rizomélico); d) múltiplos pontos de calcificações na topografia das epífises dos ossos longos e em carpo e tarso; e) articulações de joelho e cotovelo dispostos em flexão; f) regiões metafisárias levemente alargadas (Figuras 2 e 3); g) calcificações puntiformes nas junções costovertebrais e regiões pediculares dos corpos vertebrais (incluso sacro); h) fenda coronal nos corpos das vértebras L2 e L5; i) fenda sagital nos corpos das vértebras T9 e L2 (Figura 4).
A paciente recebeu alta hospitalar com diagnóstico clínico-radiológico de CDP e foram fornecidas informações e orientações aos pais sobre o caso.
DISCUSSÃO As displasias ósseas se caracterizam por alterações no crescimento e desenvolvimento cartilagíneo, ósseo e do remodelamento destes(7), principalmente as formas autossômica dominante (doença de Conradi-Hünnermann) e recessiva (rizomélica). A forma dominante da CDP é a forma mais freqüente e está relacionada a defeito na biossíntese enzimática dos peroxissomas. O quadro clínico varia de uma forma muito grave com catarata, encurtamento assimétrico dos ossos longos, escoliose, lesões cutâneas tipo ictiose e fácies achatada com sela nasal alargada e plana, a uma forma bem mais leve(6). Há melhor expectativa de vida, embora o óbito fetal precoce também ocorra. Os que sobrevivem ao primeiro ano em geral apresentam expectativa de vida e desenvolvimento mental normais. As epífises puntiformes em geral calcificam-se tardiamente e apresentam um aspecto dismórfico, porém algumas podem adquirir aspecto praticamente normal(3). A forma rizomélica da CDP é de origem autossômica recessiva, caracterizada por defeito funcional dos peroxissomas, resultando em deficiência enzimática, na qual há diminuição na síntese de plasmalogênio, redução da oxidação de ácido fitânico e presença de uma enzima peroxissômica hepática não processada (inativa), a 3-oxacil-Coa-tiolase(5). Atualmente, o diagnóstico desta forma de CDP é feito por meio das características clínicas compatíveis com a síndrome, associadas a achados bioquímicos que incluem dosagens séricas do ácido fitânico, pesquisa de síntese de plasmalogênio em cultura de fibroblastos(5), bem como no interior dos eritrócitos e nível dos ácidos graxos de cadeia longa no plasma(2). O ácido fitânico plasmático encontra-se elevado, e a síntese de plasmalogênio nos fibroblastos e seu conteúdo nos eritrócitos estão reduzidos(2,4,8,9). O nível sérico dos ácidos graxos de cadeia longa costuma estar normal(2). O estudo cromossômico denota mutação no gene PEX7, sendo 50% delas no alelo L292ter(5). Nenhum destes testes, bioquímico ou genético, foi realizado no caso em análise. Esta variante da CDP é rara, com incidência estimada em 1:100.000(6) e com apenas 72 casos descritos na literatura até 1995(10). As principais características descritas na literatura são micromelia rizomélica (encurtamento proximal dos membros) simétrica e acentuada; calcificações puntiformes e alterações da ossificação em metáfises e epífises de ossos longos; calcificações puntiformes e fissuras coronais em vértebras de coluna torácica e lombar; microcefalia e atraso de crescimento, retardo psicomotor, espasticidade e óbito precoce(5, 6,11). A presença de fendas vertebrais, zona longitudinal radiotransparente observada no perfil, antes descritas como invariáveis nos casos de CDP rizomélica, não se mostrou presente em três de cinco casos avaliados por Wardinsky et al.(2) e em outros casos na literatura, não sendo obrigatoriamente necessária ao diagnóstico(2). Outras características têm sido descritas com freqüência variável, entre elas ictiose, catarata, mobilidade articular restrita, dificuldade de sucção e deglutição, alopécia, deficiência auditiva e visual, convulsões, hipoplasia dos nervos ópticos, cifoescoliose e espinha bífida(2,5,12,13). Os pacientes comumente apresentam fácies com micrognatia, hipoplasia malar, ponte nasal achatada e ponta bulbosa, aparentando uma face achatada. Ao contrário das demais formas de CDP, a rizomélica tem prognóstico ruim, com infecções de repetição e óbito nos dois primeiros anos de vida(2,4–6). O diagnóstico diferencial inclui as outras causas de CDP, síndrome de Keutel, síndrome de Zellweger, síndrome de Smith-Lemn-Opitz, doença de Refsum neonatal e clássica, adrenoleucodistrofia neonatal, lúpus neonatal, trissomia do 21 ou 18, síndrome alcoólica fetal, infecções congênitas e uso materno de fenitoína ou dicumarínicos durante a gestação(4–6,10). A paciente apresentava ictiose, focos salteados de alopécia, pele frouxa, pescoço encurtado, fácies achatada com nariz em sela, articulação de joelho e cotovelo em flexão permanente, além de encurtamento dos membros, microcefalia e micrognatia, e história de dificuldade de deglutição, características clínicas que corroboram o diagnóstico de CDP rizomélica. Uma característica comum da CDP rizomélica é a presença de fissuras coronais em corpos vertebrais. Neste caso, foram identificadas fissuras coronais nos corpos vertebrais de L2 e L5, além de encurtamento proximal simétrico bilateral dos ossos dos membros inferiores e superiores com pontos de calcificação na topografia das epífises dos ossos longos, no carpo e no tarso. Essas fissuras são decorrentes de fusão deficiente das metades anterior e posterior dos corpos vertebrais, que ocorre por volta do quarto mês de gestação(2,5,14); já as calcificações puntiformes freqüentemente observadas são devidas à degeneração da cartilagem que se processa de modo progressivo e representada por condrócitos de núcleo picnótico e citoplasma eosinófilo acompanhados por ossificação(14). Deficiências auditiva e/ou visual não foram observadas, embora sejam achados de freqüência variável na bibliografia consultada. Outro achado característico deste tipo de condrodisplasia é a microcefalia, que se mostrou bastante acentuada, haja vista seus índices estarem muito abaixo do normal. Com base nos dados apresentados, inferimos o diagnóstico como CDP forma recessiva, em virtude da exuberância do quadro clínico e radiológico; no entanto, a confirmação diagnóstica por meio de testes bioquímicos e genéticos não foi possível de ser realizada. É importante observar que pacientes com diagnóstico de CDP rizomélica devem ser acompanhados ambulatorialmente, pois apesar de inexistência de tratamento específico atualmente, muitas das manifestações clínicas podem não estar presentes no momento do diagnóstico, mas aparecerem na evolução do caso, tais como alopécia, ictiose e catarata. Outras tendem a desaparecer com a idade, como as calcificações puntiformes, sem deixar deformidades ósseas(5). Atualmente, a paciente ora em análise encontra-se em acompanhamento ambulatorial nesta instituição.
REFERÊNCIAS 1. Mortier GR, Messiaen LM, Espeel M, et al. Chondrodysplasia punctata with multiple congenital anomalies: a new syndrome? Pediatr Radiol 1998; 28:790–793. [ ] 2. Wardinsky TD, Pagon RA, Powell BR, et al. Rhizomelic chondrodysplasia punctata and survival beyond one year: a review of the literature and five case reports. Clin Genet 1990;38:84–93. [ ] 3. Labrunie E, Pereira LF, Guimarães RR. Displasia epifisária punctata – relato de caso. Radiol Bras 1999;23:161–164. [ ] 4. Kumada S, Hayashi M, Kenmochi J, et al. Lethal form of chondrodysplasia punctata with normal plasmalogen and cholesterol biosynthesis. Am J Med Genet 2001;98:250–255. [ ] 5. Pascolat G, Zindeluk JL, Abraão KC, Rodrigues FM, Guedes CIM. Condrodisplasia puntiforme forma rizomélica – relato de caso. J Pediatr 2003; 79:189–192. [ ] 6. Omobono E, Goetsch W. Chondrodysplasia punctata (the Conradi-Hünnermann syndrome). A clinical case report and review of the literature. Minerva Pediatr 1993;45:117–121. [ ] 7. Tristán López J, Zafra de la Rosa G, Manzano C, Hernández Correa P, López Zendejas G. Síndrome de condrodisplasia punctata rizomélica. Informe de un caso. Rev Mex Ped 1993;60:59–61. [ ] 8. Ikegawa S, Ohashi H, Ogata T, et al. Novel and recurrent EBP mutations in X-linked dominant chondrodysplasia punctata. Am J Med Genet 2001;94:300–305. [ ] 9. Heymans HS, Oorthuys JW, Nelck G, Wanders RJ, Schutgens RB. Rhizomelic chondrodysplasia punctata: another peroxisomal disorder [letter]. N Engl J Med 1985;313:187–188. [ ] 10. Fourie DT. Chondrodysplasia punctata: case report and literature review of patients with heart lesions. Pediatr Cardiol 1995;16:247–250. [ ] 11. Spranger JW, Opitz JM, Bidder U. Heterogeneity of chondrodysplasia punctata[abstract]. Humangenetik 1971;11:190–212. [ ] 12. Agamanolis DP, Novak RW. Rhizomelic chondrodysplasia punctata: report of a case with review of the literature and correlation with other peroxisomal disorders. Pediatr Pathol Lab Med 1995; 15:503–513. [ ] 13. Jansen V, Sarafoglou K, Rebarber A, Greco A, Genieser NB, Wallerstein R. Chondrodysplasia punctata, tibial-metacarpal type in a 16 week fetus. J Ultrasound Med 2000;19:719–722. [ ] 14. Poulos A, Sheffield L, Sharp P, et al. Rhizomelic chondrodysplasia punctata: clinical, pathologic, and biochemical findings in two patients. J Pediatr 1988;113:685–690. [ ]
Recebido para publicação em 20/4/2005.
* Trabalho realizado no Hospital de Base Dr. Ary Pinheiro – Universidade Federal de Rondônia, Porto Velho, RO. |
|

{kind=link}
Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554