Radiologia Brasileira - Publicação Científica Oficial do Colégio Brasileiro de Radiologia
AMB - Associação Médica Brasileira CNA - Comissão Nacional de Acreditação
 Vol. 40 nº 1 - Jan. /Feb. of 2007
Vol. 40 nº 1 - Jan. /Feb. of 2007
|
CASE REPORT
|
|
Rhizomelic chondrodysplasia punctata: a case report and brief literature review |
|
|
Autho(rs): Sizenildo da Silva Figueirêdo, Jobelino Sousa de Araújo, José Eduardo Marini Kozan, Nilton César Lima dos Santos, Valessa Tanganeli |
|
|
Keywords: Chondrodysplasia punctata, Bone dysplasia, Peroxisomal disorders |
|
|
Abstract:
IProfessor of the Discipline of Imaginology at Faculdade de Medicina da Universidade Federal de Rondônia, Titular Member of Colégio Brasileiro de Radiologia e Diagnóstico por Imagem
INTRODUCTION Epiphyseal stippling is a radiological sign present in severalbone dysplasias, and is associated with different metabolic,teratogenic and chromosomal disorders. These punctatecalcifications may result in delayed endochondral ossificationprocess, growth deficiency and deformity of the bonesinvolved(1). Amongst the causes of epiphysealstippling, special importance is given to chondrodysplasiapunctata (CDP), a condition correlated with phenotypic variationsand determined by the type of genetic transmission. CDP corresponds to a heterogeneous group of dysplasiascharacterized by punctate calcifications in cartilage (mainly theepiphyseal ones), frequently associated with limbs shortening,cataracts, ichthyosis, alopecia, nervous system alterations,mental and growth deficiencies(2–5). High rate of stillbirth or mortality during the first year oflife is reported, as a result of associated anomalies orintercurrent diseases(3). Today, the CDPdiagnosis is made by means of clinical analysis concomitant withbiochemical and radiological findings. Although quite evidentclinical and radiological characteristics might determine anaccurate diagnosis, other diseases should be taken intoconsideration because of findings similarity. The CPD family includes the autosomal dominant form(Conradi-Hünnermann syndrome), the autosomal recessive(rhizomelic), the X-linked recessive (CDPX1) and dominant (CDPX2or Conradi-Hünnermann-Happlesyndrome)(5). The autosomal forms result from a peroxisomal metabolicdisorder(4,6), the X-linked dominant (CDPX2) iscaused by defects in cholesterol biosynthesis pathway, and theX-linked recessive (CDPX1) results from defects in arysulfataseE(4). Peroxisomes are organelles present in thecytoplasm of several tissular cells (especially in the liver,kidneys and fibroblasts), containing a complex enzymatic systemwith an array of functions such as: several chemicals oxidationand production of H2O2; biliary acid,cholesterol and cell plasmalogen biosynthesis; â-oxidationof fatty acids with long carbon chains; lysine metabolism andglyosylate synthesis/metabolism(6). A case of CDP is presented, with a description of the two mainvariations (autosomal recessive and dominant) based on a briefreview of the existent literature about this diseaseetiopathogenesis, clinical-radiological and laboratorymanifestations.
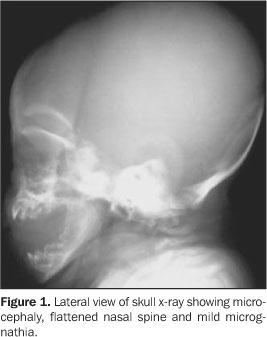
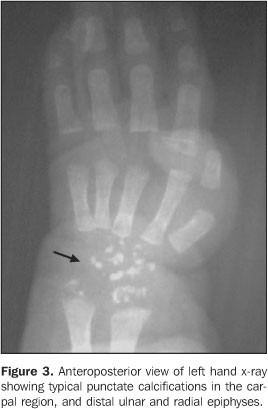
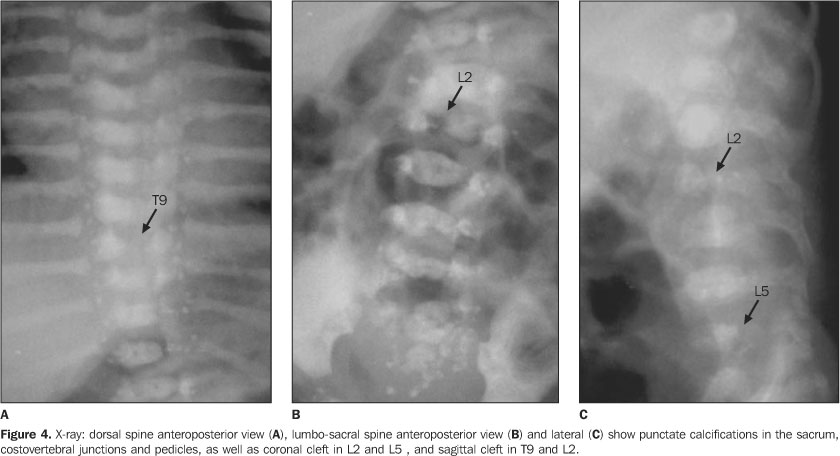
CASE REPORT Female, two-month-old infant, has been referred to thePediatrics Service of Hospital de Base Ary Pinheiro, Porto Velho,RO, Brazil, because of tachypnea and history of frequent chokingsafter breastfeeding, as well as evident anatomical alterations inface and limbs. She was kept in a thermoregulated incubator withcirculating O2, during 11 days. Preterm-born from consanguineous parents (primiparous mother),the patient has presented a series of clinical and radiologicalfindings suggesting a CDP diagnosis. Her mother denied a historyof drugs or alcohol abuse or exposure to warfarin (known asteratogenic causes of punctate epiphyses). Clinically, the patient presented with ichthyotic skinchanges, irregular foci of alopecia, shortening of permanentlyflexed and spastic limbs, micrognathia; flattened nose, smallchest with restricted expansion, cephalhematoma, besides thealready mentioned tachypnea and groaning. Cardio-pulmonaryauscultation showed mild bibasal crepitation and absence ofabdominal alterations. Results from somatometry were thefollowing: weight 1.755 g, height 37 cm, both cephalic andthoracic perimeters 25 cm (below the fifth percentile). Routinelaboratory tests as well as those performed for investigatingcongenital infections were normal. On radiographic studies, we have observed: a) micrognathia; b) flattened nasal spine (Figure 1); c) symmetrical, bilateral, proximal shortening of upper and lower limbs (rhizomelic pattern); d) multiple punctate calcifications in the epiphyseal cartilage of long bones, ankle and carp; e) flexed knee and elbow joints; f) slightly enlarged metaphyseal regions (Figures 2 and 3); g) punctate calcifications in costochondral junctions and vertebral bodies pedicles (including the sacrum); h) coronal clefts of L2 and L5 vertebral bodies; i) sagittal clefts of T9 and L2 vertebral bodies (Figure 4).
The patient was released from the hospital withclinical-radiological diagnosis of CDP. Parents were giveninformation and guidance on the case.
DISCUSSION Bone dysplasias are characterized by changes in growth,cartilaginous and bone development, as well as in boneremodeling(7), mainly the autosomical dominant(Conradi-Hünnermann syndrome) and recessive (rhizomelic)forms of the disease. The CDP dominant form is most frequent and is related to adefect in peroxisomal enzyme biosynthesis. The clinical picturemay range from mild disease to an extremely severe condition,with cataracts, asymmetrical shortening of long bones, scoliosis,ichthyotic-type skin lesions, and flattened facies with broadnasal bridge(6). There is some life expectancy,although early fetal death also occurs. Infants who survivebeyond the first year usually present normal life expectancy andmental development. In general, punctate epiphyses presentdelayed calcification with a dysmorphic aspect, but some of themmay progress to a practically normalaspect(3). The rhizomelic CDP is of autosomal recessive origin,characterized by a peroxisomes functional defect resulting in anenzymatic deficiency where there is a decrease in the plasmalogensynthesis, decrease in phytanic acid oxidation and presence of aunprocessed (inactive), the3-oxacyl-Coa-thiolase(5). Currently this form of CDP is diagnosed through clinicalfeatures compatible with the syndrome associated with biochemicalfindings including phytanic acid serum levels, screening ofplasmalogen synthesis on culturedfibroblasts(5), as well as in erythrocytes, andplasmatic level of fatty acids with long carbonchain(2). The plasmatic level of phytanic acidis high, and the plasmalogen synthesis in fibroblast anderythrocytes is reduced(2,4,8,9). Usually, theserum level of fatty acids with long carbon chain isnormal(2). The chromosomal study demonstratesPEX7 mutation, 50% of them in the L292terallele(5). None of these biochemical or genetictests has been performed in the present case. This CDP variant israre, with an estimate 1:100,000 incidence(6),and with only 72 cases reported in the literature up to1995(10). The main characteristics described in the literature aresymmetric and severe rhizomelic micromelia (proximal shorteningof limbs); punctate calcifications and long bones metaphyseal andepiphyseal ossification changes; punctate calcifications andcoronal clefts in vertebral bodies of the thoracic and lumbarspine; microcephaly and growth retardation, psychomotor delay,spasticity and precocious death(5,6,11). Thepresence of vertebral clefts, radiotransparent, longitudinal zoneobserved in the lateral view, previously described as invariablein cases of rhizomelic CDP, has not been present in three of fivecases analyzed by Wardinsky et al.(2) and inother cases reported in the literature, and is not obligatorilynecessary for the diagnosis(2). Other characteristics have been described with a variablefrequency, among them ichthyosis, cataracts, restricted jointmobility, sucking and deglutition difficulty, alopecia, auditiveand visual dificiencies, seizures, optic nerves hypoplasia,kyphoscoliosis and cleft spine(2,5,12,13).Patients usually present facies with micrognathia, malarhypoplasia, flattened nasal bridge and bulbous nose, withflattened face appearance. In contrast to the other forms of CDP,the rhizomelic CDP presents a poor prognosis, with repetitioninfections and death in the first two years oflife(2,4–6). The differential diagnosis includes other causes for CDP,Keutel syndrome, Zellweger syndrome, Smith-Lemn-Opitz syndrome,classical and neonatal Refsum disease, neonataladrenoleucodystrophy, neonatal lupus, trisomy 21 or 18, fetalalcoholic syndrome, congenital infections, and maternal use ofcoumarin-like compounds or phenitoine duringgestation(4–6,10). The patient presented ichthyosis, irregular foci of alopecia,feeble skin, shortened neck, flattened facies with saddle nose,permanently flexed knee and elbow joints, besides limbsshortening, microcephaly and micrognathia and history ofdeglutition difficulty, clinical characteristics corroboratingthe diagnosis of rhizomelic CDP. An usual characteristic of therhizomelic CDP is the presence of coronal clefts in vertebralbodies. In the present case, coronal clefts in L2 and L5vertebral bodies were identified, besides proximal, symmetricalbilateral shortening of upper and lower limbs bones with punctatecalcifications on the long bones epiphyses, carp and ankle. Theseclefts are a result of poor fusion of anterior and posteriorhalves of vertebral bodies, occurring around the fourth month ofgestation(2,5,14); on the other hand,frequently observed punctate calcifications are caused by aprogressive cartilage degeneration represented by chondrocyteswith a pycnotic nucleus and eosinophil cytoplasm followed byossification(14). Auditive and/or visual dificiencies have been observed,although being found with variable frequency in the referredliterature. However, another typical finding in this type ofchondrodysplasia is microcephaly, which has shown to be quiteaccentuated, considering the values very below than normal. Based on the data presented by this study, we have inferredthe diagnosis as recessive CDP considering the exuberance of theclinical-radiological picture; however, we could not confirm thediagnosis by means of biochemical and genetic tests. It is important to note that patients with diagnosis ofrhizomelic CDP should undergo ambulatorial follow-up, as, inspite of the current inexistence of specific treatment, manyclinical manifestations, like alopecia, ichthyosis and cataracts,might not be present at the moment of the diagnosis, showing upwith the progress of the disease. Other manifestations, likepunctate calcifications, tend to disappear with aging, withoutresulting in bone deformities(5). Presently,the patient is being kept in ambulatorial follow-up in ourinstitution.
REFERENCES 1. Mortier GR, Messiaen LM, Espeel M, et al. Chondrodysplasia punctata with multiple congenital anomalies: a new syndrome? Pediatr Radiol 1998; 28:790–793. [ ] 2. Wardinsky TD, Pagon RA, Powell BR, et al. Rhizomelic chondrodysplasia punctata and survival beyond one year: a review of the literature and five case reports. Clin Genet 1990;38:84–93. [ ] 3. Labrunie E, Pereira LF, Guimarães RR. Displasia epifisária punctata – relato de caso. Radiol Bras 1999;23:161–164. [ ] 4. Kumada S, Hayashi M, Kenmochi J, et al. Lethal form of chondrodysplasia punctata with normal plasmalogen and cholesterol biosynthesis. Am J Med Genet 2001;98:250–255. [ ] 5. Pascolat G, Zindeluk JL, Abraão KC, Rodrigues FM, Guedes CIM. Condrodisplasia puntiforme forma rizomélica – relato de caso. J Pediatr 2003; 79:189–192. [ ] 6. Omobono E, Goetsch W. Chondrodysplasia punctata (the Conradi-Hünnermann syndrome). A clinical case report and review of the literature. Minerva Pediatr 1993;45:117–121. [ ] 7. Tristán López J, Zafra de la Rosa G, Manzano C, Hernández Correa P, López Zendejas G. Síndrome de condrodisplasia punctata rizomélica. Informe de un caso. Rev Mex Ped 1993;60:59–61. [ ] 8. Ikegawa S, Ohashi H, Ogata T, et al. Novel and recurrent EBP mutations in X-linked dominant chondrodysplasia punctata. Am J Med Genet 2001;94:300–305. [ ] 9. Heymans HS, Oorthuys JW, Nelck G, Wanders RJ, Schutgens RB. Rhizomelic chondrodysplasia punctata: another peroxisomal disorder [letter]. N Engl J Med 1985;313:187–188. [ ] 10. Fourie DT. Chondrodysplasia punctata: case report and literature review of patients with heart lesions. Pediatr Cardiol 1995;16:247–250. [ ] 11. Spranger JW, Opitz JM, Bidder U. Heterogeneity of chondrodysplasia punctata[abstract]. Humangenetik 1971;11:190–212. [ ] 12. Agamanolis DP, Novak RW. Rhizomelic chondrodysplasia punctata: report of a case with review of the literature and correlation with other peroxisomal disorders. Pediatr Pathol Lab Med 1995; 15:503–513. [ ] 13. Jansen V, Sarafoglou K, Rebarber A, Greco A, Genieser NB, Wallerstein R. Chondrodysplasia punctata, tibial-metacarpal type in a 16 week fetus. J Ultrasound Med 2000;19:719–722. [ ] 14. Poulos A, Sheffield L, Sharp P, et al. Rhizomelic chondrodysplasia punctata: clinical, pathologic, and biochemical findings in two patients. J Pediatr 1988;113:685–690. [ ]
Received April 20, 2005.
* Study developed at Hospital de Base Dr. Ary Pinheiro – Universidade Federal de Rondônia, Porto Velho, RO, Brazil. |
|


{kind=link}
Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554