Radiologia Brasileira - Publicação Científica Oficial do Colégio Brasileiro de Radiologia
AMB - Associação Médica Brasileira CNA - Comissão Nacional de Acreditação
 Vol. 41 nº 3 - Maio / Jun. of 2008
Vol. 41 nº 3 - Maio / Jun. of 2008
|
ARTIGO DE REVISÃO
|
|
|
|
|
Autho(rs): C. Isabela S. Silva, Nestor L. Müller |
|
|
Descritores: Pulmão, Doença do colágeno, Pneumonia intersticial, Hipertensão pulmonar, Tomografia computadorizada |
|
|
Resumo:
INTRODUÇÃO As doenças do colágeno (colagenoses) correspondem a um grupo de doenças com características autoimunes que acometem vários órgãos, incluindo o pulmão(1,2). As manifestações intratorácicas são freqüentes, podendo ser assintomáticas ou sintomáticas e com graus variados de gravidade. O padrão e a freqüência de comprometimento intratorácico dependem do tipo específico de doença do colágeno, que pode envolver um ou vários compartimentos simultaneamente, tais como alvéolo, interstício, vasos, tecido linfático, vias aéreas e pleura(1-7). As manifestações pulmonares mais comuns incluem as pneumonias intersticiais difusas(1,2,6) e a hipertensão pulmonar(1,2,8), que em conjunto representam as principais causas de mortalidade e morbidade nesses pacientes(1,2). É importante salientar que as doenças pulmonares em pacientes com colagenose podem ser decorrentes não apenas da própria doença de base, mas também da terapêutica instituída, ocasionando danos pulmonares medicamentosos ou infecções bacterianas ou por germes oportunistas, como Pneumocystis jiroveci, e micobacterioses atípicas por causa da imunossupressão(1,2). As doenças do colágeno que mais comumente cursam com comprometimento intersticial e que possuem maior significância clínica incluem: artrite reumatóide, esclerose sistêmica progressiva, lúpus eritematoso sistêmico, dermatopolimiosite, doença mista do tecido conjuntivo e síndrome de Sjögren(1,2,6). A radiografia de tórax representa o método de imagem mais utilizado na avaliação inicial das manifestações intratorácicas das doenças do colágeno. A radiografia, entretanto, apresenta baixas sensibilidade e especificidade. A tomografia computadorizada de alta resolução (TCAR) do tórax é o método de escolha para a avaliação das alterações pulmonares nas doenças do colágeno, ao oferecer melhor correlação clínico-histológica, estadiamento da gravidade da doença, avaliação de prognóstico e progressão, bem como diagnóstico diferencial(1). O objetivo deste artigo é revisar as manifestações intratorácicas e o diagnóstico diferencial principal das doenças do colágeno na TCAR.
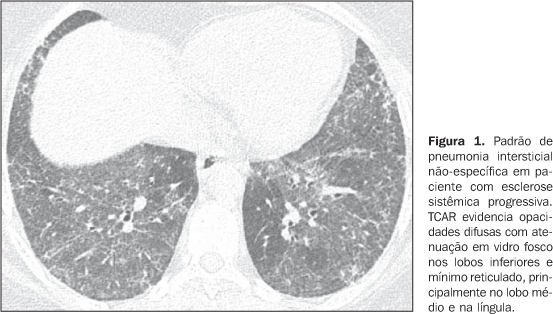
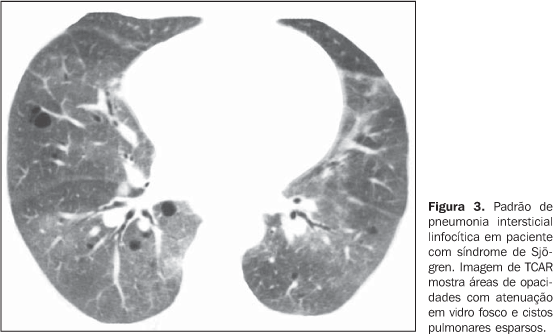
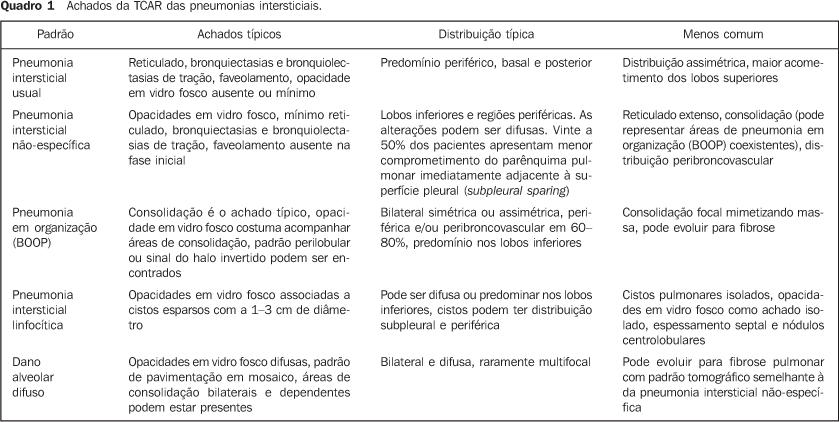
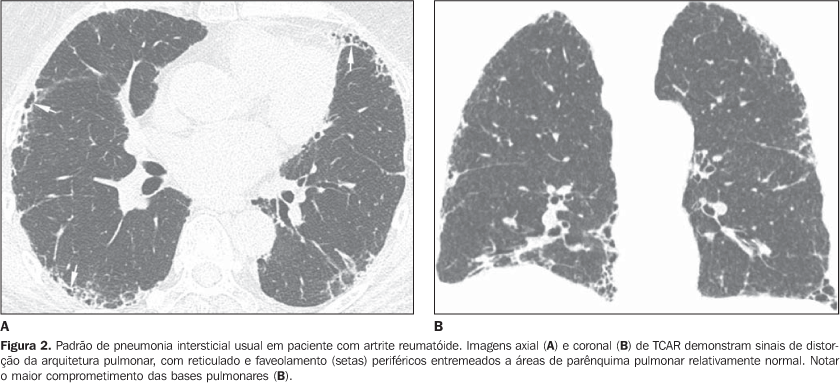
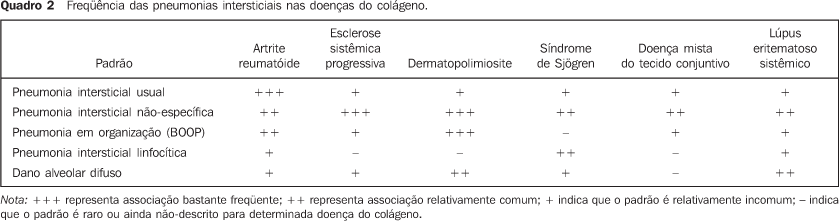
PRINCIPAIS MANIFESTAÇÕES INTRATORÁCICAS DAS DOENÇAS DO COLÁGENO 1. Pneumonias intersticiais difusas As pneumonias intersticiais representam as manifestações pulmonares mais comuns das colagenoses(1,2). As doenças do colágeno podem associar-se a praticamente todos os padrões de pneumonias intersticiais difusas, podendo inclusive progredir para fibrose pulmonar(1-3,6). A classificação histológica é semelhante à das pneumonias intersticiais de natureza idiopática e inclui: a) padrões de evolução crônica - pneumonia intersticial usual, pneumonia intersticial não-específica e pneumonia intersticial linfocítica; b) padrão de evolução subaguda - pneumonia em organização (também conhecida como bronquiolite obliterante com pneumonia em organização —BOOP); c) padrão de injúria pulmonar de evolução aguda - dano alveolar difuso(3,9,10). Os achados tomográficos das pneumonias intersticiais em pacientes com doenças do colágeno também são semelhantes aos descritos para pacientes com doença idiopática (Quadro 1)(1-3,9,11). As doenças do colágeno mais comumente associadas a pneumonias intersticiais fibrosantes são a artrite reumatóide, a esclerose sistêmica progressiva (esclerodermia) e a dermatopolimiosite. Em geral, o padrão de pneumonia intersticial mais comum em pacientes com doença do colágeno é o de pneumonia intersticial não-específica (Figura 1)(1,2). Em pacientes com artrite reumatóide, todavia, apesar de ainda controverso, o padrão de pneumonia intersticial usual (Figura 2) parece ser o mais freqüente(1,7). Já a pneumonia intersticial linfocítica, embora rara, é mais comum em pacientes com síndrome de Sjögren (Figura 3)(1,2,12), e o padrão de dano alveolar agudo é mais freqüente em pacientes com dermatopolimiosite e lúpus eritematoso sistêmico. O Quadro 2 mostra a freqüência de cada padrão de pneumonia intersticial encontrada nas principais doenças do colágeno discutidas nesta revisão.
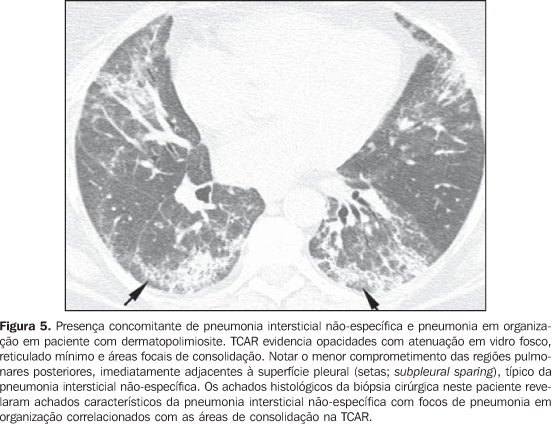
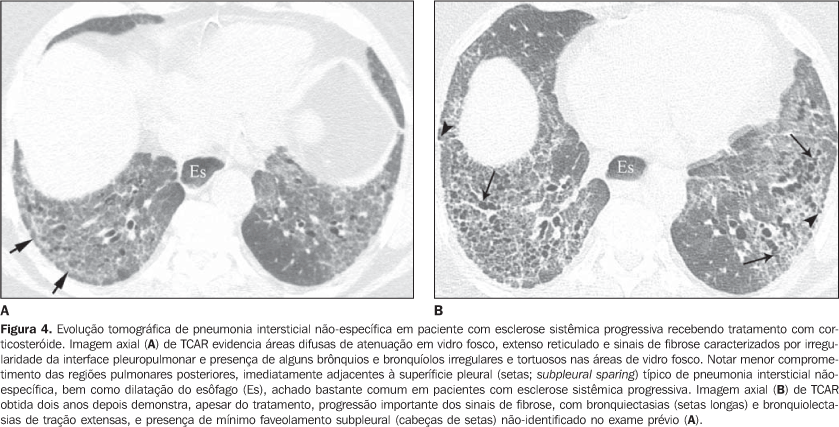
As pneumonias intersticiais de evolução crônica nas doenças do colágeno possuem, em geral, melhor prognóstico do que as de natureza idiopática, com melhor tempo de sobrevida em cinco anos (Figura 4)(13). Entretanto, casos de evolução desfavorável em pacientes com doença do colágeno e comprometimento pulmonar intersticial sem resposta ao tratamento não são incomuns. Semelhante à forma idiopática, o padrão de pneumonia intersticial não-específica na doença do colágeno tem melhor prognóstico do que o padrão de pneumonia intersticial usual(13). A presença concomitante de mais de um padrão de pneumonia intersticial, principalmente a associação entre pneumonia intersticial não-específica e pneumonia em organização (BOOP) não é rara nas colagenoses, sendo mais freqüente em pacientes com dermatopolimiosite e doença mista do tecido conjuntivo (Figura 5). O padrão de dano alveolar difuso geralmente traduz mau prognóstico, tanto em pacientes com doença idiopática quanto em pacientes com doença do colágeno(10).
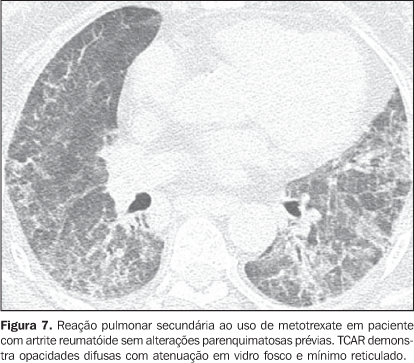
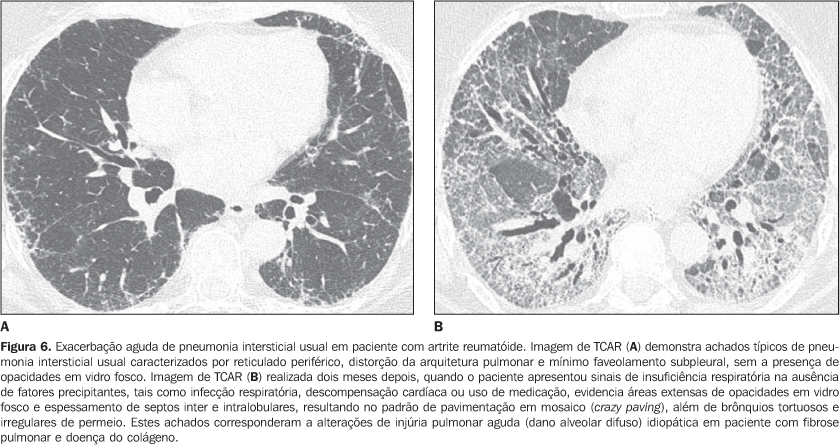
As pneumonias intersticiais podem preceder a apresentação clínica da doença do colágeno de três meses até cinco anos, geralmente com padrão de pneumonia intersticial não-específica(14) ou, agudamente, com padrão de dano alveolar difuso. Pacientes com doença do colágeno, principalmente artrite reumatóide, esclerodermia e dermatopolimiosite, podem evoluir com exacerbação aguda da pneumonia intersticial fibrosante(15). A injúria aguda do pulmão mais freqüentemente manifesta-se na histologia com padrão de dano alveolar difuso superposto a fibrose pulmonar já estabelecida (geralmente pneumonia intersticial usual ou pneumonia intersticial não-específica)(15) . Os achados de TCAR da exacerbação aguda da pneumonia intersticial de evolução crônica em pacientes com doença do colágeno consistem de áreas difusas de opacidades em vidro fosco com ou sem a presença concomitante de espessamento liso dos septos inter e intralobulares, resultando no padrão de pavimentação em mosaico (crazy paving), associadas a sinais de fibrose da pneumonia intersticial preexistente (distorção da arquitetura pulmonar, bronquiectasias e bronquiolectasias de tração, reticulado e faveolamento) (Figura 6)(15,16). Áreas de consolidação focal ou bilaterais dependentes podem ser encontradas. O diagnóstico diferencial principal na TCAR de áreas difusas de opacidades em vidro fosco em paciente com doença do colágeno e pneumonia intersticial fibrosante deve incluir infecções oportunistas (principalmente por Pneumocystis) e reação pulmonar medicamentosa (Figura 7)(1-3). Consolidação pode corresponder a pneumonia em organização ou processo infeccioso (por bactérias, micobactérias ou fungos).
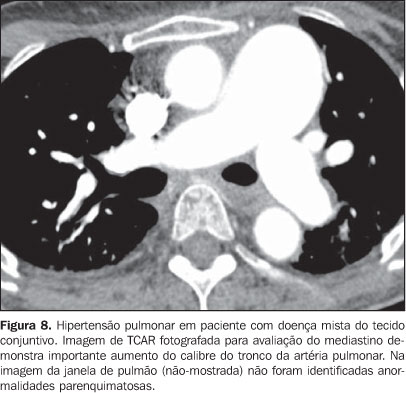
Nos pacientes fumantes e com doença do colágeno deve-se ressaltar que pneumonias intersticiais relacionadas ao tabagismo também podem ser encontradas, tais como a bronquiolite respiratória com doença intersticial pulmonar e a pneumonia intersticial descamativa(2,9). 2. Doença pulmonar vascular A vasculopatia pulmonar nas doenças do colágeno geralmente manifesta-se como hipertensão pulmonar, hemorragia pulmonar difusa (capilarite) ou vasculite de pequenos e médios vasos(2,8). A doença pulmonar venoclusiva e a hemangiomatose pulmonar capilar, apesar de raras, já foram descritas em alguns pacientes com doença do colágeno, como esclerodermia e lúpus eritematoso sistêmico(8,17). A hipertensão pulmonar na doença do colágeno pode resultar de uma variedade de mecanismos fisiopatológicos diferentes, que incluem: perda da rede capilar pulmonar como conseqüência da doença intersticial fibrosante; arteriopatia pulmonar intrínseca com remodelação vascular e hipertensão arterial pulmonar; disfunção diastólica ventricular esquerda com hipertensão venosa pulmonar; e doença tromboembólica crônica associada com hipercoagulabilidade e anticorpos antifosfolípides (pacientes com lúpus eritematoso sistêmico)(8,18). Pode ser achado isolado (Figura 8) ou ocorrer na presença de pneumonia intersticial fibrosante. A incidência da hipertensão pulmonar nas doenças do colágeno é variável, dependendo do tipo de doença do colágeno, da presença concomitante de doença intersticial e do método diagnóstico utilizado(8,18). A hipertensão pulmonar é mais comum na esclerodermia (aproximadamente 25% dos pacientes), no lúpus eritematoso sistêmico (5-10%) e na doença mista do tecido conjuntivo(8,18).
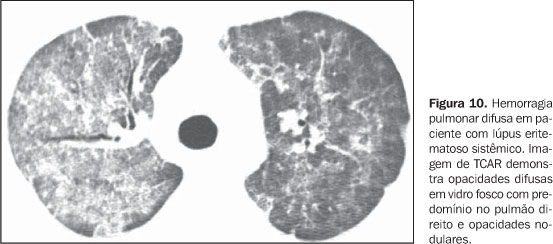
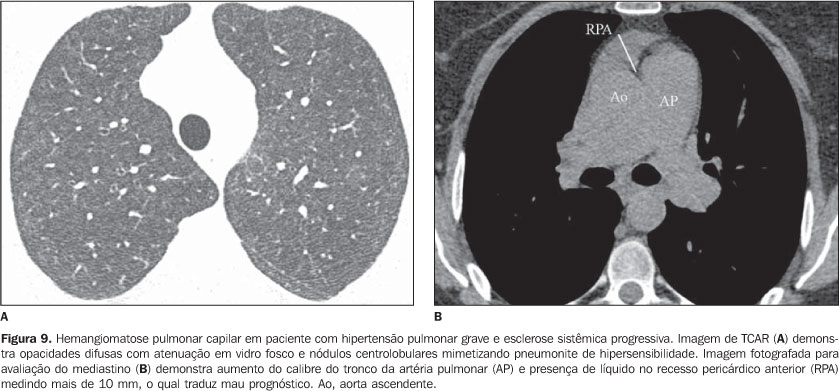
O aumento do diâmetro do tronco da artéria pulmonar (> 2,9 cm) (Figura 8), das artérias pulmonares principais e segmentares representam os achados característicos da hipertensão pulmonar na TCAR(3). Calcificação das paredes das artérias podem sem identificadas em pacientes com hipertensão grave e de longa data, bem como trombos, principalmente em pacientes com lúpus eritematoso sistêmico e anticorpos antifosfolípides. O padrão de perfusão em mosaico pode estar presente. Raramente nódulos centrolobulares e opacidades em vidro fosco difusas ou esparsas são identificados na TCAR (Figura 9). Estas alterações podem ser multifatoriais e serem resultantes da própria hipertensão pulmonar (proliferação das pequenas artérias musculares, congestão vascular passiva crônica que pode levar a ingurgitamento do leito capilar, depósito de granulomas de colesterol, áreas focais de hemorragia) ou estarem associadas à presença de hemangiomatose pulmonar capilar (Figura 9) (proliferação multifocal exagerada de capilares dentro das paredes alveolares). A presença concomitante de espessamento de septos interlobulares em pacientes com hipertensão pulmonar pode ser secundária a insuficiência cardíaca direita ou, menos comumente, a obliteração venosa encontrada na doença pulmonar venoclusiva. Os achados principais na TCAR de hemorragia pulmonar difusa (capilarite) na doença do colágeno consistem de opacidades em vidro fosco difusas (Figura 10), padrão de pavimentação em mosaico, consolidações esparsas, e nódulos centrolobulares ou áreas lobulares de opacidades em vidro fosco(3). O diagnóstico diferencial tomográfico principal inclui infecções pulmonares, principalmente pneumonia por Pneumocystis, dano alveolar difuso (Figura 6) e reação pulmonar medicamentosa (Figura 7)(1).
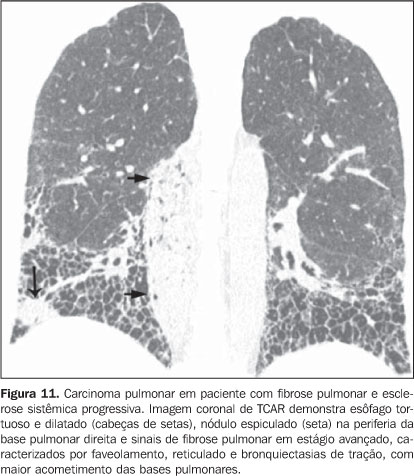
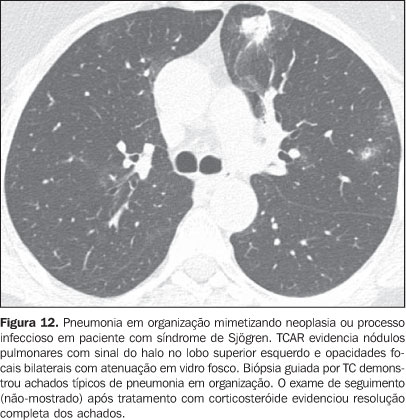
3. Nódulos ou massas pulmonares O diagnóstico diferencial de nódulos ou massas pulmonares em pacientes com doença do colágeno é extenso e deve incluir processo infeccioso geralmente oportunista (Nocardia, Cryptococcus,Aspergillus, Histoplasma, micobactérias atípicas), neoplásico (carcinoma, linfoma), metabólico (amiloidose) e imunológico (nódulos reumatóides)(19). A seguir, resumimos as principais causas de nódulos e massas pulmonares na TCAR de pacientes com colagenose. a) Nódulos reumatóides - Raros, mas podem ser encontrados em pacientes do sexo masculino com artrite reumatóide, nódulos subcutâneos e fator reumatóide positivo(1,20). Medem entre 0,5 cm e 7 cm de diâmetro, podendo ser únicos ou múltiplos com centro necrótico circundado por histiócitos e leucócitos na histologia. Raramente se desenvolvem na pleura ou pericárdio. Os nódulos reumatóides podem sofrer cavitação (até 50%) e assim causarem hemoptise ou pneumotórax ou tornarem-se infectados(19-21). Resolução espontânea pode ocorrer. A presença de calcificação é extremamente incomum(19). Uma rara associação de nódulos reumatóides em pacientes com doença reumatóide e pneumoconiose (principalmente a pneumoconiose dos trabalhadores de carvão) é designada de síndrome de Caplan(19). b) Linfoma pulmonar - Mais comum em pacientes com síndrome de Sjögren, pode ser encontrado também em pacientes com artrite reumatóide(19,22). Na TCAR podem apresentar-se como nódulos (geralmente com mais que 1,0 cm de diâmetro), consolidação ou, raramente, opacidades em vidro fosco(12,22). c) Amiloidose - A forma nodular da amiloidose pode ocorrer em pacientes com síndrome de Sjögren e pneumonia intersticial linfocítica(22,23). Os nódulos podem ser isolados ou múltiplos, apresentar calcificação e localizar-se no parênquima pulmonar ou no interior dos cistos pulmonares. A forma difusa da amiloidose pulmonar é rara, mas pode ser encontrada em pacientes com artrite reumatóide e síndrome de Sjögren(12,19,22,24). Na TCAR a amiloidose pulmonar difusa é caracterizada por micronódulos difusos e espessamento de septos interlobulares. d) Carcinoma pulmonar - Estudos demonstram incidência aumentada de carcinoma pulmonar em pacientes com doença do colágeno e doença intersticial fibrosante concomitante(1,19), mas esta relação não é ainda bem estabelecida(25,26). A incidência estimada de neoplasia (incluindo câncer de pulmão) em pacientes com esclerodermia é de 3,6% a 10,7%(27). Recentemente, observou-se que a incidência de câncer de pulmão em pacientes com esclerodermia não está relacionada à presença concomitante de fibrose pulmonar, subtipo da esclerodermia e status imunológico; por outro lado, pacientes fumantes e com esclerodermia apresentam risco sete vezes maior para desenvolver câncer de pulmão do que não-fumantes(28). Na TCAR o carcinoma pulmonar pode manifestar-se como nódulo espiculado ou massa pulmonar (Figura 11). O diagnóstico diferencial principal inclui nódulo reumatóide (na artrite reumatóide), processos infecciosos (principalmente fúngico), amiloidose focal e linfoma pulmonar (principalmente em pacientes com síndrome de Sjögren) e pneumonia em organização (BOOP) localizada (Figura 12).
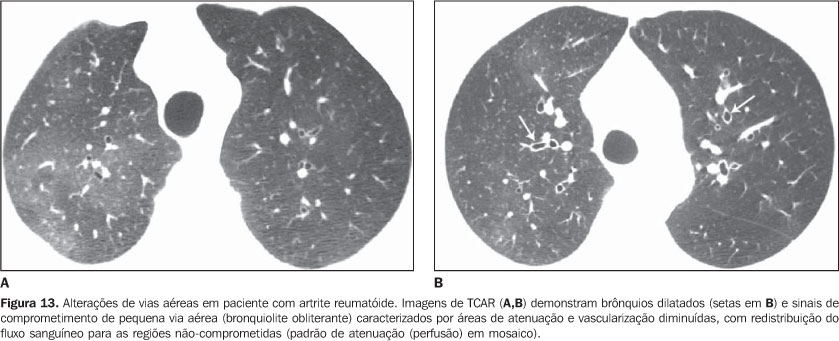
4. Consolidação pulmonar difusa Em pacientes com lúpus eritematoso sistêmico pode corresponder a pneumonite lúpica ou hemorragia pulmonar difusa (Figura 10)(1,19). Reação pulmonar medicamentosa deve sempre ser considerada no diagnóstico diferencial. A presença de consolidação predominando nas regiões pulmonares dependentes, principalmente quando associada a nódulos centrolobulares, pode traduzir pneumonia aspirativa, achado bastante comum em pacientes com esclerodermia e dermatopolimiosite, por causa da dismotilidade esofágica(19,29). 5. Via aérea a) Grande via aérea - bronquiectasias Mais comum na artrite reumatóide (até 30%), podendo também ser encontrada na síndrome de Sjögren e no lúpus eritematoso sistêmico(1,3,19). Na TCAR é mais freqüente nos lobos inferiores e do tipo cilíndrica (Figura 13)(7). O fator causal da bronquiectasia em pacientes com doença do colágeno é multifatorial, podendo ser decorrente de processo infeccioso de repetição (principalmente na artrite reumatóide), comprometimento da pequena via aérea ou ressecamento da via aérea (síndrome de Sjögren)(1-3,7). b) Pequena via aérea - bronquiolites - Bronquiolite obliterante (constritiva): ocorre principalmente em mulheres com artrite reumatóide e, menos comumente, em pacientes com síndrome de Sjögren(1-3). Representa destruição da parede bronquiolar por tecido de granulação e eventual substituição dos bronquíolos por tecido fibrótico. Os achados principais da TCAR incluem áreas de diminuição da atenuação e vascularização pulmonar, com ou sem redistribuição do fluxo sanguíneo para as regiões não-comprometidas, resultando no padrão de atenuação (perfusão) em mosaico (Figura 13)(3,7). Na TCAR expiratória observa-se aprisionamento aéreo. Brônquios dilatados (bronquiectasias) (Figura 13) e de paredes espessadas podem ser encontrados(3,7). Deve-se ressaltar que a bronquiolite obliterante também pode ser resultante do tratamento com penicilamina em pacientes com artrite reumatóide(19). A hipertensão pulmonar também pode resultar no padrão de perfusão (atenuação) em mosaico na TCAR, entretanto, não costuma apresentar aprisionamento aéreo na TCAR expiratória e, diferentemente da bronquiolite obliterante, observa-se aumento do calibre da artéria pulmonar. - Bronquiolite folicular: representa hiperplasia de tecido linfóide bronquiolocêntrico, mais comumente encontrada na artrite reumatóide, síndrome de Sjögren e esclerodermia(1-3,30). A TCAR pode ser normal ou mostrar nódulos centrolobulares e nódulos peribroncovasculares(31). O diagnóstico diferencial principal na TCAR inclui processo infeccioso e aspiração (mais comum nas regiões pulmonares dependentes). - Bronquiolite exsudativa (celular): geralmente de natureza infecciosa, caracterizada na TCAR como nódulos centrolobulares ou opacidades nodulares ramificadas (árvore em brotamento) com distribuição bilateral e assimétrica(3). 6. Outras manifestações intratorácicas das doenças do colágeno As doenças do colágeno também podem estar associadas a outras anormalidades intratorácicas, resumidas a seguir. a) Linfonodomegalia Aumento de linfonodos mediastinais é bastante comum em pacientes com doença do colágeno. Na grande maioria dos casos a linfonodomegalia está associada à presença concomitante de doença pulmonar intersticial fibrosante, sendo encontrada em cerca de 70% dos pacientes com esclerodermia(19,32). Usualmente, os linfonodos medem até 1,5 cm de diâmetro no menor eixo e acometem somente uma ou duas estações. Linfonodos hilares e mediastinais aumentados podem, todavia, ser encontrados em qualquer paciente com doença do colágeno, inclusive na ausência de doença intersticial, e apresentar causas variadas, tais como: sarcoidose, infecção, linfoma, metástase, resposta inflamatória à presença de refluxo ou pneumonia aspirativa(19,32). b) Alterações da pleura e pericárdio O acometimento pleural (derrame ou espessamento) é bastante freqüente na doença do colágeno, principalmente no lúpus eritematoso sistêmico e na artrite reumatóide(1,19,33). A presença concomitante de derrame pericárdico é comum. Mais da metade dos pacientes com lúpus eritematoso sistêmico desenvolve, no curso da doença, derrame pleural, geralmente bilateral e de aspecto seroso ou serossanguinolento(33). O derrame pleural, bem como o pericárdico, podem também ser decorrentes do uso de procainamida e hidralazina em pacientes com lúpus eritematoso sistêmico(19). Na artrite reumatóide o espessamento pleural é encontrado em até 70% dos pacientes. O derrame pleural, por outro lado, é identificado em cerca de 5% dos pacientes com artrite ativa, sendo geralmente unilateral e de volume pequeno ou moderado(19). Em muitos pacientes as alterações pericárdicas (derrame pericárdico, aderência pericárdica e pericardite fibrinosa ou fibrosa) são assintomáticas (até 50%)(34). Derrame pericárdico freqüentemente associado a derrames pleurais é uma das manifestações intratorácicas mais comuns do lúpus eritematoso sistêmico. O derrame pericárdico, de modo geral, tem bom prognóstico. Entretanto, é importante salientar que a presença de derrame pericárdico em pacientes com doença do colágeno, quando associada a hipertensão pulmonar (definida por meio de ecocardiografia), principalmente quando se identifica líquido no recesso pericárdico anterior medindo mais do que 10 mm (Figura 9B), geralmente traduz mau prognóstico(34,35). c) Disfunção diafragmática Incomum, mas pode ser encontrada em pacientes com lúpus eritematoso sistêmico e ocasionalmente em pacientes com dermatopolimiosite(1,3). Resulta em dispnéia, padrão restritivo nas provas de função pulmonar na ausência de alterações pulmonares e elevação diafragmática com atelectasias basais adjacentes. d) Dismotilidade esofágica Comum em pacientes com esclerodermia e dermatopolimiosite(19). Dilatação esofágica assintomática pode ser identificada em 40% a 80% dos pacientes com esclerodermia (Figura 4)(29). Na TCAR o diâmetro coronal da luz do esôfago em pacientes com esclerodermia é de 1,2 cm a 4,0 cm (média de 2,3 cm)(29). A dismotilidade esofágica pode levar à pneumonia aspirativa.
CONCLUSÃO As manifestações intratorácicas das doenças do colágeno são bastante comuns e de grande importância clínica. As principais manifestações pulmonares incluem as pneumonias intersticiais difusas e a hipertensão pulmonar, que, em conjunto, representam as principais causas de mortalidade e morbidade nesses pacientes. O padrão e a freqüência do comprometimento parenquimatoso, vascular, de via aérea pleural e pericárdico, bem como as alterações diafragmáticas e esofágicas dependem do tipo específico de doença do colágeno. É fundamental o conhecimento dos vários padrões de acometimento que podem ser encontrados na TCAR desses pacientes, assim como a correlação clínico-radiológica. Não-raramente, as alterações intratorácicas das colagenoses podem ser multicompartimentais ou serem decorrentes da terapêutica instituída ou da imunossupressão.
REFERÊNCIAS 1. Devaraj A, Wells AU, Hansell DM. Computed tomographic imaging in connective tissue diseases. Semin Respir Crit Care Med. 2007;28: 389-97. [ ] 2. Woodhead F, Wells AU, Desai SR. Pulmonary complications of connective tissue diseases. Clin Chest Med. 2008;29:149-64, vii. [ ] 3. Tanaka N, Newell JD, Brown KK, et al. Collagen vascular disease-related lung disease: high-resolution computed tomography findings based on the pathologic classification. J Comput Assist Tomogr. 2004;28:351-60. [ ] 4. Tansey D, Wells AU, Colby TV, et al. Variations in histological patterns of interstitial pneumonia between connective tissue disorders and their relationship to prognosis. Histopathology. 2004; 44:585-96. [ ] 5. Primack SL, Müller NL. Radiologic manifestations of the systemic autoimmune diseases. Clin Chest Med. 1998;19:573-86, vii. [ ] 6. Lamblin C, Bergoin C, Saelens T, et al. Interstitial lung diseases in collagen vascular diseases. Eur Respir J Suppl. 2001;32:69S-80. [ ] 7. Tanaka N, Kim JS, Newell JD, et al. Rheumatoid arthritis-related lung diseases: CT findings. Radiology. 2004;232:81-91. [ ] 8. Hoeper MM. Pulmonary hypertension in collagen vascular disease. Eur Respir J. 2002;19:571-6. [ ] 9. Lynch DA, Travis WD, Muller NL, et al. Idiopathic interstitial pneumonias: CT features. Radiology. 2005;236:10-21. [ ] 10. Won Huh J, Soon Kim D, Keun Lee C, et al. Two distinct clinical types of interstitial lung disease associated with polymyositis-dermatomyositis. Respir Med. 2007;101:1761-9. [ ] 11. Desai SR, Veeraraghavan S, Hansell DM, et al. CT features of lung disease in patients with systemic sclerosis: comparison with idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia. Radiology. 2004;232:560-7. [ ] 12. Parambil JG, Myers JL, Lindell RM, et al. Interstitial lung disease in primary Sjögren syndrome. Chest. 2006;130:1489-95. [ ] 13. Park JH, Kim DS, Park IN, et al. Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am J Respir Crit Care Med. 2007;175:705-11. [ ] 14. Sato T, Fujita J, Yamadori I, et al. Non-specific interstitial pneumonia; as the first clinical presentation of various collagen vascular disorders. Rheumatol Int. 2006;26:551-5. [ ] 15. Silva CI, Müller NL, Fujimoto K, et al. Acute exacerbation of chronic interstitial pneumonia: high-resolution computed tomography and pathologic findings. J Thorac Imaging. 2007;22:221-9. [ ] 16. Horoupian N, English J, Müller NL. Accelerated deterioration of usual interstitial pneumonia with acute development of honeycomb cysts in rheumatoid arthritis. J Thorac Imaging. 2004;19:127-30. [ ] 17. Kishida Y, Kanai Y, Kuramochi S, et al. Pulmonary venoocclusive disease in a patient with systemic lupus erythematosus. J Rheumatol. 1993; 20:2161-2. [ ] 18. Handa T, Nagai S, Miki S, et al. Incidence of pulmonary hypertension and its clinical relevance in patients with interstitial pneumonias: comparison between idiopathic and collagen vascular disease associated interstitial pneumonias. Intern Med. 2007;46:831-7. [ ] 19. Rockall AG, Rickards D, Shaw PJ. Imaging of the pulmonary manifestations of systemic disease. Postgrad Med J. 2001;77:621-38. [ ] 20. Tanoue LT. Pulmonary involvement in collagen vascular disease: a review of the pulmonary manifestations of the Marfan syndrome, ankylosing spondylitis, Sjögrens syndrome, and relapsing polychondritis. J Thorac Imaging. 1992;7:62-77. [ ] 21. Kobayashi T, Satoh K, Ohkawa M, et al. Multiple rheumatoid nodules with rapid thin-walled cavity formation producing pneumothorax. J Thorac Imaging. 2005;20:47-9. [ ] 22. Jeong YJ, Lee KS, Chung MP, et al. Amyloidosis and lymphoproliferative disease in Sjögren syndrome: thin-section computed tomography findings and histopathologic comparisons. J Comput Assist Tomogr. 2004;28:776-81. [ ] 23. Desai SR, Nicholson AG, Stewart S, et al. Benign pulmonary lymphocytic infiltration and amyloidosis: computed tomographic and pathologic features in three cases. J Thorac Imaging. 1997;12: 215-20. [ ] 24. Sumiya M, Ohya N, Shinoura H, et al. Diffuse interstitial pulmonary amyloidosis in rheumatoid arthritis. J Rheumatol. 1996;23:933-6. [ ] 25. Pearson JE, Silman AJ. Risk of cancer in patients with scleroderma. Ann Rheum Dis. 2003;62:697-9. [ ] 26. Hill CL, Nguyen AM, Roder D, et al. Risk of cancer in patients with scleroderma: a population based cohort study. Ann Rheum Dis. 2003;62: 728-31. [ ] 27. Wooten M. Systemic sclerosis and malignancy: a review of the literature. South Med J. 2008;101: 59-62. [ ] 28. Pontifex EK, Hill CL, Roberts-Thomson P. Risk factors for lung cancer in patients with scleroderma: a nested case-control study. Ann Rheum Dis. 2007;66:551-3. [ ] 29. Bhalla M, Silver RM, Shepard JA, et al. Chest CT in patients with scleroderma: prevalence of asymptomatic esophageal dilatation and mediastinal lymphadenopathy. AJR Am J Roentgenol. 1993;161:269-72. [ ] 30. Hayakawa H, Sato A, Imokawa S, et al. Bronchiolar disease in rheumatoid arthritis. Am J Respir Crit Care Med. 1996;154:1531-6. [ ] 31. Howling SJ, Hansell DM, Wells AU, et al. Follicular bronchiolitis: thin-section CT and histologic findings. Radiology. 1999;212:637-42. [ ] 32. Wechsler RJ, Steiner RM, Spirn PW, et al. The relationship of thoracic lymphadenopathy to pulmonary interstitial disease in diffuse and limited systemic sclerosis: CT findings. AJR Am J Roentgenol. 1996;167:101-4. [ ] 33. Kim JS, Lee KS, Koh EM, et al. Thoracic involvement of systemic lupus erythematosus: clinical, pathologic, and radiologic findings. J Comput Assist Tomogr. 2000;24:9-18. [ ] 34. Fischer A, Misumi S, Curran-Everett D, et al. Pericardial abnormalities predict the presence of echocardiographically defined pulmonary arterial hypertension in systemic sclerosis-related interstitial lung disease. Chest. 2007;131:988-92. [ ] 35. Baque-Juston MC, Wells AU, Hansell DM. Pericardial thickening or effusion in patients with pulmonary artery hypertension: a CT study. AJR Am J Roentgenol. 1999;172:361-4. [ ]
Recebido para publicação em 5/5/2008. Aceito, após revisão, em 27/5/2008.
* Trabalho realizado na University of British Columbia, Vancouver General Hospital, Vancouver, Canadá |
|

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554