Radiologia Brasileira - Publicação Científica Oficial do Colégio Brasileiro de Radiologia
AMB - Associação Médica Brasileira CNA - Comissão Nacional de Acreditação
 Vol. 55 nº 2 - Mar. / Abr. of 2022
Vol. 55 nº 2 - Mar. / Abr. of 2022
|
ENSAIO PICTÓRICO
|
|
|
|
|
Autho(rs): André Felix Pedri1,a; Marcelo dos Santos Guedes1,2,b; Cláudio Campi de Castro2,c |
|
|
Descritores: Erros inatos do metabolismo/classificação; Encefalopatias metabólicas congênitas/classificação; Diagnóstico por imagem/tendências; Tomografia computadorizada; Ressonância magnética. |
|
|
Resumo: INTRODUÇÃO
As doenças neurometabólicas hereditárias (DNMHs) representam um desafio diagnóstico. Constituem um grupo de doenças raras e geralmente graves, porém, algumas têm possibilidade terapêutica. Há variabilidade genética, mas a maioria é de herança autossômica recessiva. Os quadros clínico e radiológico, a depender da fase do diagnóstico, podem ser bastante variáveis e superponíveis, o que torna a elucidação mais complexa(1). Vários estudos já oferecem classificações, algumas de aplicação prática difícil(1). O avanço tecnológico das últimas décadas, em especial dos métodos de imagem, tem propiciado melhora na sensibilidade e na especificidade da avaliação das doenças do sistema nervoso central. Ressalta-se o papel da ressonância magnética (RM) e suas técnicas avançadas no estudo das DNMHs(1,2). Baseados nos aspectos atrás descritos, os autores detectaram a necessidade de elaborar um estudo fundamentado exclusivamente nos achados radiológicos das DNMHs, agrupando-os em um único trabalho e subdivididos em grupos com fins didáticos. Assim, este é o objetivo: classificar as DNMHs de acordo com achados radiológicos, a fim de permitir um guia prático de consulta. Este é um estudo retrospectivo, com dados obtidos em uma rede de hospitais. Foram revisados exames de tomografia computadorizada multidetectores (TCMD) e/ou RM do sistema nervoso central, de pacientes na fase etária predominantemente pediátrica, submetidos a exames entre janeiro de 2010 e agosto de 2020. Foram considerados apenas os casos cujos diagnósticos definitivos foram estabelecidos por meio de resultados anatomopatológicos, laboratoriais e/ou genéticos, com 74 casos confirmados. Em seguida, tais casos foram subdivididos em grupos de acordo com os achados radiológicos. Foram caracterizados 10 tipos de padrões neurorradiológicos distintos que permitiram a divisão dos casos nos seguintes subgrupos: macrocrania, presença de cistos, localização do acometimento da substância branca, realce pós-contraste, presença de calcificações, envolvimento dos núcleos da base e tálamos, restrição à difusão, alterações vasculares, envolvimento de nervos cranianos e padrão de metabólitos na espectroscopia de prótons por RM (EPRM). MACROCRANIA Macrocrania se refere ao aumento do perímetro cefálico. Fazem parte das DNMHs que podem cursar com macrocrania: leucoencefalopatia megaloencefálica com cistos subcorticais (LMC), doença de Alexander, ácido-2-hidroglutárico, mucopolissacaridoses e doença de Canavan. Apesar de esse achado, isoladamente, não permitir o diagnóstico definitivo, reduz as hipóteses e, quando em associação com outros achados, pode auxiliar na definição do diagnóstico(16) (Figura 1). 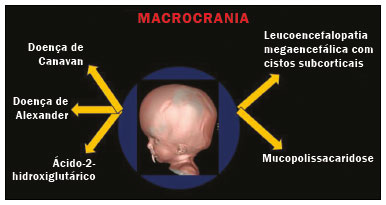 Figura 1. Macrocrania em DNMHs. Reconstrução 3D volume rendering, obtida de imagens axiais de TCMD do crânio, exemplifica aumento do volume do crânio e suas possíveis causas (setas). PRESENÇA DE CISTOS O achado de imagens de aspecto cístico pode ser observado em uma porcentagem considerável de casos de DNMHs(1,2). A origem dos cistos é ainda incerta, se bem que, em sua maioria, deva corresponder a degeneração da substância branca, aumento do espaço periencefálico ou dilatação dos espaços perivasculares. A localização, o número e o tamanho dos chamados cistos podem auxiliar no diagnóstico diferencial. As DNMHs que podem cursar com cistos são: LMC (cistos subcorticais que têm predomínio em polos temporais), acidúria glutárica tipo 1 (alargamento de fissuras silvianas), leucodistrofia com cistos e calcificações (síndrome de Labrune), mucopolissacaridoses (dilatação de espaços perivasculares), leucoencefalopatia cavitante progressiva (cistos periventriculares, com acometimento do corpo caloso), leucoencefalopatia com substância branca evanescente (cistos periventriculares) e síndrome de Aicardi. Há outras menos frequentes, como doença de Alexander (cistos frontais), doença de Tay-Sachs, síndrome de Zellweger (cistos germinolíticos), leucoencefalopatia com envolvimento do tronco e medula e lactato aumentado (doença caracterizada por lesões de substância branca, tratos no tronco cerebral e região posterior da medula espinhal, que, devido à heterogeneidade de sinal da substância branca pode demonstrar, em alguns casos, presença de imagens de aspecto cístico), síndrome de Lowe, deficiência do cofator do molibdênio, galactosemia e doença de Leigh(16) (Figura 2). 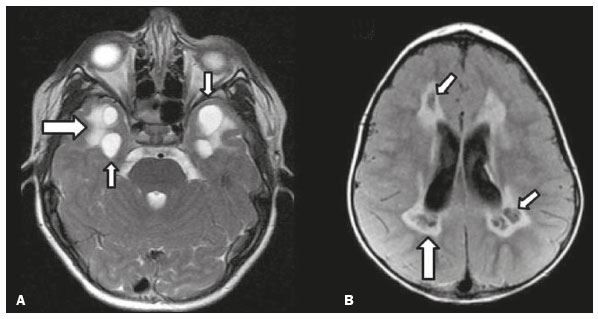 Figura 2. Presença de cistos em DNMHs. Imagens de RM de crânio. A: Axial TSE pesada em T2 mostra hipersinal da substância branca compatível com leucoencefalopatia (seta maior), com cistos subcorticais de permeio (setas menores) em regiões temporais, em caso de LMC. B: Axial FLAIR de outro caso mostra hipersinal da substância branca periventricular supratentorial bilateral compatível com leucoencefalopatia (seta maior), com cistos de permeio (setas menores), em caso de leucoencefalopatia cavitante progressiva. LOCALIZAÇÃO DO ACOMETIMENTO DA SUBSTÂNCIA BRANCA Um dos achados radiológicos mais significativos para DNMHs é o local do acometimento da substância branca. Na quase totalidade dessas doenças ocorre envolvimento da substância branca, na maioria dos casos, supratentorial. Pode haver, também, acometimento do tronco cerebral e do cerebelo(1,2). O acometimento pode ser confluente, multifocal, periventricular, subcortical, difuso, anterior, posterior, ou ambos, mas geralmente é bilateral(1,2) (Figura 3). Ressalta-se, entretanto, que há variações e que, a depender da fase evolutiva da doença no momento da realização do estudo, este tipo de avaliação pode ser prejudicado, pela tendência de as DNMHs apresentarem, em suas fases finais ou crônicas, sinais radiológicos superponíveis. Em relação a tal acometimento, observa-se também o achado radiológico denominado padrão tigroide ou pele de leopardo, resultado de desmielinização com preservação de algumas fibras perivenulares, encontrado na leucodistrofia metacromática (LDM) e, às vezes, na doença de Krabbe e na leucoencefalopatia com substância branca evanescente(13) (Figura 4). 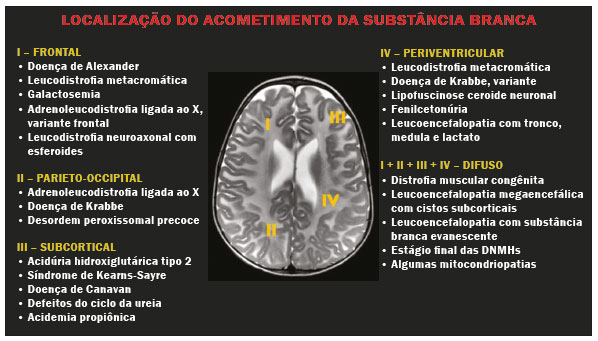 Figura 3. Esquema do acometimento da substância branca e suas possíveis causas em DNMHs, com base em imagem axial TSE pesada em T2 de crânio (números de I a IV). 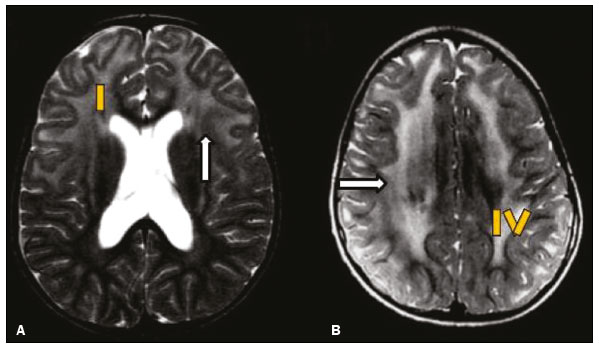 Figura 4. Acometimento da substância branca em DNHMs. Imagens axiais de RM de crânio, correlacionadas com o esquema da Figura 3. A: TSE pesada em T2 mostra lesões confluentes de hipersinal na substância branca frontal em caso de doença de Alexander (seta e número I). B: TSE pesada em T2 mostra hipersinal da substância branca periventricular superior bilateral com padrão tigroide em caso de LDM (seta e número IV). REALCE PÓS-CONTRASTE Algumas DNMHs apresentam realce parenquimatoso pelo meio de contraste, particularmente: adrenoleucodistrofia ligada ao X (ALD-X), doença de Alexander, leucoencefalopatia cavitante progressiva, doença de Krabbe e síndrome de Labrune. Entretanto, os representantes mais característicos deste achado radiológico são: doença de Alexander com o realce nodular periventricular anterior, configurando o sinal radiológico da orelha de coelho, e a ALD-X, esta com realce periférico dividido nas três zonas de Schaumburg(1,2,6,7) (Figura 5). 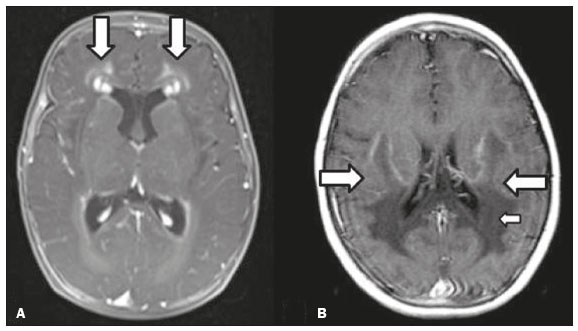 Figura 5. Realce pelo meio de contraste em DNMHs. Imagens de RM pós-contraste de crânio. A: Axial SE pesada em T1 identifica realce nodular periventricular anterior, configurando o sinal radiológico da orelha de coelho (setas) em caso de doença de Alexander. B: Axial SE pesada em T1 de um paciente com ALD-X mostra hipossinal e realce periférico pós-contraste da substância branca parieto-occipital bilateral, caracterizando as três zonas de Schaumburg área periférica anterior, em que predomina edema vasogênico; área intermediária de desmielinização ativa com realce (setas maiores); e área central de gliose (seta menor). PRESENÇA DE CALCIFICAÇÕES Há DNMHs que apresentam calcificações. A detecção deste achado radiológico é extremamente relevante para o diagnóstico. Ressalta-se a importância da TCMD e de sequências de suscetibilidade (SWI e GRET2*) na RM na detecção de calcificações, pela alta sensibilidade e especificidade. As DNMHs que podem apresentar calcificações, estas predominando nas regiões nucleocapsulares, são: síndrome de Labrune, síndrome de Aicardi, doença de Fabry, síndrome MELAS, síndrome de Kearns-Sayre e, eventualmente, doença de Leigh. Deve-se considerar, ainda, ALD-X quando houver calcificações de substância branca(13,7,8) (Figura 6). 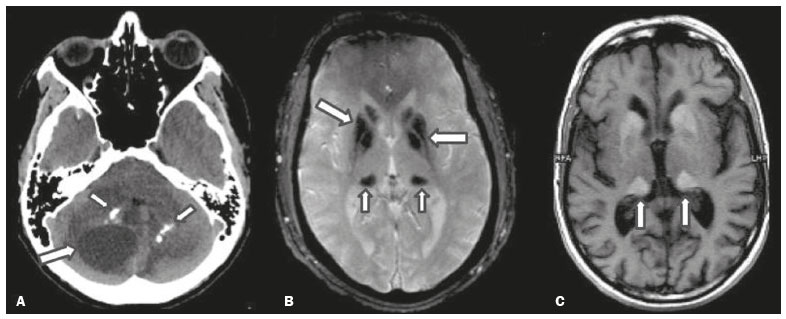 Figura 6. DNMHs com calcificações. A: Imagem axial de TCMD de crânio sem contraste mostra imagens hipodensas císticas (seta maior) associadas a calcificações (setas menores) em hemisférios cerebelares, em caso de síndrome de Labrune. B: Imagem axial de RM T2* de outro paciente identifica focos hipointensos bilaterais em núcleos da base (setas maiores) e no pulvinar dos tálamos sinal do pulvinar (setas menores) em caso de doença de Fabry. C: Imagem axial SE pesada em T1 sem contraste, do mesmo paciente da Figura B, exibe hipersinal nas regiões de hipossinal em T2*, inclusive no pulvinar dos tálamos (setas). ENVOLVIMENTO DOS NÚCLEOS DA BASE E TÁLAMOS Outros achados, além de calcificações, podem ser observados nos núcleos da base e tálamos em DNMHs, como hiperdensidade na TCMD e, às vezes, hipersinal em T1 na RM em tálamos, em doença de Krabbe, mucopolissacaridoses e doença de Tay-Sachs. Hipersinal em T2 e FLAIR na RM, particularmente nos núcleos caudados, globos pálidos e putâmens, pode aparecer em acidúria glutárica tipo 1, doença de Leigh e síndrome de Kearns-Sayre. Há, ainda, doenças que cursam com o chamado sinal do tálamo preto nas imagens de RM TSE pesadas em T2: lipofuscinose, gangliosidoses (GM1 e GM2), fucosidose, doença de Wilson e POLR3 (Figura 7). Atrofia dos núcleos caudados e putâmens pode ser observada na doença de Huntington e fase final de algumas DNMHs(13,8,9). 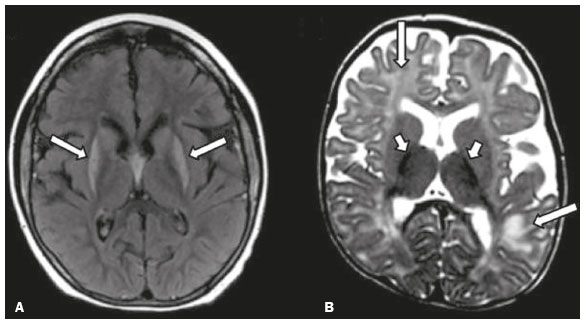 Figura 7. Acometimento dos núcleos da base e tálamos em DNMHs. Imagens de RM de crânio. A: Axial FLAIR mostra hipersinal bilateral e simétrico em núcleos da base, particularmente em putâmens, com redução de volume local (setas), em caso de doença de Leigh. B: Axial TSE ponderada em T2 mostra leve espessamento e hipossinal em T2 dos tálamos (setas menores), associado a hipersinal difuso da substância branca periventricular e subcortical (setas maiores) em caso de gangliosidose (GM1). RESTRIÇÃO À DIFUSÃO As DNMHs podem cursar, principalmente em suas fases iniciais ou de atividade, com edema parenquimatoso, seja vasogênico e/ou citotóxico. Se ocorrer edema citotóxico, podem apresentar restrição à difusão nas imagens de RM. As que mais frequentemente cursam com restrição à difusão são: doença da urina do xarope de bordo, hiperglicenemia não cetótica, mitocondriopatias, particularmente doença de Leigh e síndrome de Kearns-Sayre, doença de Krabbe, LDM, distúrbios do ciclo da ureia, leucoencefalopatia cavitante e, eventualmente, acidúria glutárica tipo 1, fenilcetonúria e doença de Canavan(1,2,9,10) (Figura 8). Existe ainda a possibilidade da realização da técnica de tensor de difusão, que avalia o comprometimento microestrutural da substância branca de algumas DNMHs(11). 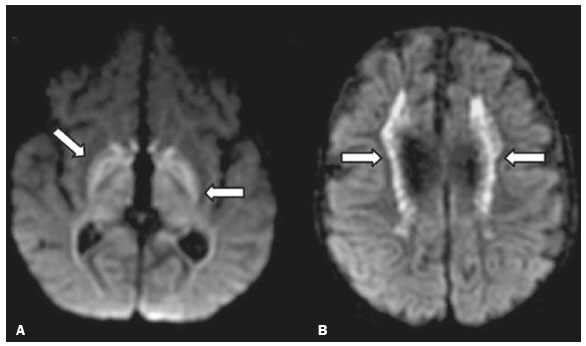 Figura 8. DNMHs com restrição à difusão. Imagens axiais de RM difusão. A: Restrição à difusão em núcleos da base e tálamos (setas) de recém-nascido com doença da urina de xarope de bordo; a hiperglicenemia não cetótica pode apresentar padrão radiológico semelhante. B: Restrição à difusão em substância branca periventricular supratentorial bilateral (setas) em caso de LDM. ALTERAÇÕES VASCULARES Há DNMHs que podem cursar com alterações vasculares. As principais são: doença de Menkes, síndrome MELAS, doença de Fabry e mucopolissacaridoses. A doença de Menkes é caracterizada por tortuosidade vascular cerebral (Figura 9), e a síndrome MELAS e a doença de Fabry, pela presença de acidente vascular encefálico e calcificações. Enfatiza-se, ainda, o sinal do shifting spread, que se refere ao aparecimento e desaparecimento, com aparecimento em outra região, de lesões isquêmicas em sítio vascular parenquimatoso na síndrome MELAS(13,8). 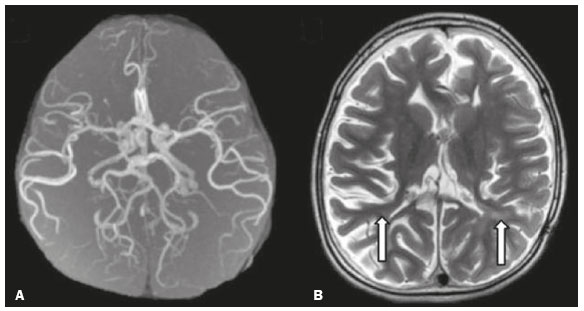 Figura 9. Alterações vasculares em DNMHs. Imagens de RM. A: Reconstrução 3D MIP de angio-RM 3DTOF de criança demonstra alongamento e tortuosidade vascular arterial. B: Axial TSE T2 do mesmo paciente mostra atrofia cerebral associada a leve hipersinal na substância branca periventricular posterior, compatível com leucoencefalopatia (setas) em caso de doença de Menkes. ENVOLVIMENTO DE NERVOS CRANIANOS O envolvimento de nervos cranianos não é frequente em DNMHs. Quando ocorre este achado, traz relevância para o diagnóstico diferencial. As doenças que podem cursar com esta alteração são: doença de Krabbe (realce de nervos cranianos, particularmente do nervo óptico) (Figura 10), LDM (neurite de nervos cranianos, à exceção do nervo óptico) e neuropatia óptica hereditária de Leber (realce de vias ópticas)(1,2,12). 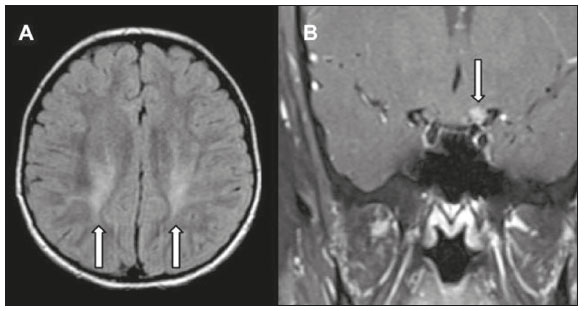 Figura 10. Envolvimento de nervos cranianos em DNMHs. Imagens de RM. Axial FLAIR (A) e coronal SE T1 pós-contraste (B) do mesmo paciente mostram hipersinal em T2 da substância branca periventricular posterior, configurando leucoencefalopatia (setas em A), associado a espessamento e realce do nervo óptico esquerdo (seta em B), em caso de doença de Krabbe. PADRÃO DE METABÓLITOS/ALTERAÇÕES NA EPRM Há DNMHs que podem mostrar alterações na EPRM. A técnica utilizada pode incluir sequências com TEs variados, sendo os mais utilizados os de 30 ms (TE curto e/ou 130 a 144 ms (TE longo), ressaltando-se o uso do TE de 270 ms na avaliação das doenças mitocondriais. As doenças mais características com alterações na EPRM são: ausência ou redução do pico de creatina (3,0 ppm, comTE curto e/ou longo) na deficiência de creatina; pico de galactitol (3,7 ppm, com TE curto) na galactosemia; elevação do pico de lactato (1,3 ppm, com TE curto, longo e com TE de até 270 ms, que neste último o lactato pode ser observado como um duplo pico acima da linha de base) em doenças mitocondriais nas fases de exacerbação clínica (doença de Leigh, síndrome MELAS, síndrome de Kearns-Sayre, etc.) e na leucoencefalopatia com acometimento do tronco e medula (Figura 11A); elevação do pico N-acetil-aspartato (2,0 ppm, de preferência com TE longo) na doença de Canavan; elevação do pico ácido-2-hidroxiglutárico (2,5 ppm, com TE curto) na acidúria glutárica tipo 2; elevação do pico da glicina (3,6 ppm, com TE longo para diferenciar glicina de mioinositol) na hiperglicenemia não cetótica; elevação dos picos de glutamina/glutamato (1,4 a 2,1 ppm, com TEs curto e longo) no distúrbio do ciclo da ureia; picos em regiões de cetoácidos e lactato (0,9 ppm e 1,3 ppm, respectivamente, com TEs curto e longo, o último em torno de 270 ms) na doença da urina do xarope de bordo; elevação do pico de fenilalanina (7,37 ppm com TE curto, em torno de 20 ms) na fenilcetonúria; e elevação do pico de succinato (2,4 ppm, com TE curto) na doença de Leigh. Doença de Krabbe, síndrome de Zellweger e LDM apresentam um padrão mais inespecífico, representado por elevação do pico de colina e, geralmente, queda do pico de N-acetil-aspartato, padrão que também pode aparecer na fase crônica ou final de outras DNMHs(1,2,9,10) (Figura 11B). 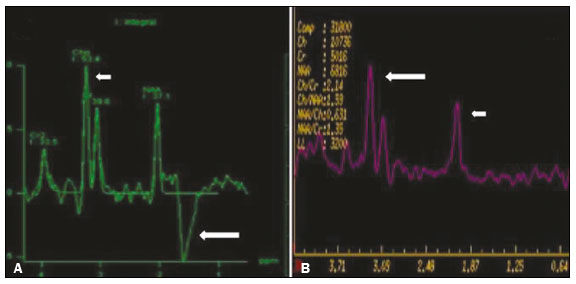 Figura 11. Alterações na EPRM em DNMHs. A: Imagem de espectroscopia pela técnica de voxel único localizado em núcleo lentiforme demonstra a presença de um pico invertido, abaixo da linha de base, no TE de 135 ms em área de lactato (seta maior) e leve elevação do pico da colina (seta menor) em caso de mitocondriopatia. B: Elevação do pico de colina (seta maior) e redução do pico de Nacetil-aspartato (seta menor), com voxel localizado na substância branca parietal, exemplificando padrão que pode estar presente nas fases crônica ou final, na maioria das leucoencefalopatias, representando um padrão inespecífico. OUTROS ACHADOS RELEVANTES Há outros sinais radiológicos em DNMHs que, quando presentes, são auxiliares no diagnóstico: malformações do desenvolvimento cortical associadas a leucoencefalopatia na síndrome de Zellweger, acidúria glutárica tipo 1 e distrofia muscular congênita; acometimento do tronco cerebral, cerebelo e da medula espinhal, poupando a substância branca supratentorial, na doença de Alexander em suas formas juvenil ou adulta; hematoma subdural na acidúria glutárica tipo 1, doença de Menkes e na doença peroxissomal não Zellweger; lesões ósseas de mãos, costelas e corpos vertebrais nas mucopolissacaridoses e, por último, acometimento cerebelar com espessamento do tendão calcâneo na xantomatose cerebrotendínea(1,2). CONCLUSÃO As DNMHs compreendem um segmento desafiador da neurorradiologia, em que muitos aspectos clínicos e de imagem se sobrepõem. Entretanto, com o emprego de achados radiológicos sugeridos na literatura e dos compilados neste ensaio, é possível estreitar o diagnóstico diferencial, aproximando o estudo por imagem do diagnóstico definitivo. REFERÊNCIAS 1. Schiffmann R, van der Knaap MS. Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology. 2009;72:7509. 2. Direnç ÖA, Alkan A. Neurometabolic diseases in children: magnetic resonance imaging and magnetic resonance spectroscopy features. Curr Med Imaging Rev. 2019;15:25568. 3. Cheon JE, Kim IO, Hwang YS, etal. Leukodystrophy in children: a pictorial review of MR imaging features. Radiographics. 2002; 22:46176. 4. Batla A, Pandey S, Nehru R. Megalencephalic leukoencephalopathy with subcortical cysts: a report of four cases. J Pediatr Neurosci. 2011;6:747. 5. Reis F, Kido RYZ, Mesquita JA, et al. Megalencephalic leukoencephalopathy with subcortical cysts (MLC) a case with clinical and magnetic resonance imaging (MRI) dissociation. Arq Neuropsiquiatr. 2015;73:1712. 6. Muralidharan CG, Tomar RPS, Aggarwal R. MRI diagnosis of Alexander disease. South African Journal of Radiology. 2012;16:1167. 7. Turk BR, Theda C, Fatemi A, et al X-linked adrenoleukodystrophy: pathology, pathophysiology, diagnostic testing, newborn screening and therapies. Int J Dev Neurosci. 2020;80:5272. 8. Sparaco M, Simonati A, Cavallaro T, et al. MELAS: clinical phenotype and morphological brain abnormalities. Acta Neuropathol. 2003;106:20212. 9. Cakmackci H, Pekcevik Y, Yis U, et al. Diagnostic value of proton MR spectroscopy and diffusion-weighted MR imaging in childhood inherited neurometabolic brain disease and review of the literature. Eur J Radiol. 2010;74:e16171. 10. Jan W, Zimmerman RA, Wang ZJ, et al. MR diffusion imaging and MR spectroscopy of maple syrup urine disease during acute metabolic decompensation. Neuroradiology. 2003;45:3939. 11. Ono SE, Carvalho Neto A, Gasparetto EL, et al. X-linked adrenoleukodystrophy: correlation between Loes score and diffusion tensor imaging parameters. Radiol Bras. 2014;47:3429. 12. Maia Jr ACM, Rocha AJ, Silva CJ, et al. Multiple cranial nerve enhancement: a new MR imaging finding in metachromatic leukodystrophy. AJNR Am J Neuroradiol. 2007;28:999. 1. Hospital Alvorada Moema, São Paulo, SP, Brasil 2. Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (HC-FMUSP), São Paulo, SP, Brasil a. https://orcid.org/0000-0002-6802-0564 b. https://orcid.org/0000-0002-6607-7006 c. https://orcid.org/0000-0002-3531-4232 Correspondência: Dr. Marcelo dos Santos Guedes Alvorada/United Health Group (UHG) Rua Conselheiro Brotero, 1486, Higienópolis São Paulo, SP, Brasil, 01232-010 E-mail: maguedes1@uol.com.br Recebido para publicação em 27/1/2021 Aceito, após revisão, em 4/4/2021 Data de publicação: 07/10/2021 |
|
Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554