Radiologia Brasileira - Publicação Científica Oficial do Colégio Brasileiro de Radiologia
AMB - Associação Médica Brasileira CNA - Comissão Nacional de Acreditação
 Vol. 53 nº 4 - Jul. / Ago. of 2020
Vol. 53 nº 4 - Jul. / Ago. of 2020
|
ARTIGO DE REVISÃO
|
|
|
|
|
Autho(rs): Marina Silva Zacariasa; Hanna Rafaela Ferreira Dalla Priab; Rafael Andrade Santiago de Oliveirac; Luis Fernando Delmonted; Fernanda Garozzo Vellonie; Giuseppe DIppolitof |
|
|
Descritores: Doenças das vias biliares; Cirrose hepática; Colangiografia; Tomografia computadorizada; Ressonância magnética. |
|
|
Resumo: INTRODUÇÃO
Colangiopatias são doenças crônicas que afetam as vias biliares com alvo nos colangiócitos. Neste artigo vamos abordar as causas não neoplásicas que compreendem um grupo heterogêneo de doenças de curso lento e progressivo, potencialmente fatais, sendo a terceira causa de transplantes hepáticos nos EUA(1,2). Apesar de suas diversas causas, essas doenças cursam com sinais e sintomas semelhantes, manifestando-se por icterícia, dor, emagrecimento e febre. De maneira análoga, os achados de imagem frequentemente se sobrepõem, o que dificulta a identificação da sua causa(3,4). Para facilitar o entendimento das diferentes formas de apresentação das colangiopatias, é necessário conhecer a anatomia das vias biliares, bem como os principais mecanismos fisiopatológicos envolvidos. Os ductos biliares intra-hepáticos são revestidos por uma única camada de células epiteliais, os pequenos colangiócitos, cuja população aumenta gradativamente até os ductos biliares extra-hepáticos (grandes colangiócitos)(5,6). Glândulas peribiliares, fontes de novos colangiócitos e de células progenitoras, acompanham a via biliar desde os ductos septais até os ductos extra-hepáticos(1,5). A irrigação dos ductos biliares é feita por ramos da artéria hepática, que formam o plexo peribiliar, enquanto a drenagem ocorre por ramos portais e veias centrolobulares(6). Na extensão da árvore biliar, os colangiócitos apresentam morfologias distintas e exercem papéis diferentes, o que em parte se deve à sua origem embriológica, sendo os ductos intra-hepáticos provenientes dos hepatoblastos e os ductos extra-hepáticos provenientes de células endodérmicas pancreáticas e duodenais(5,6). Isto também influencia a expressão de proteínas de membrana, que, em última análise, altera a forma de reação do colangiócito aos estímulos endógenos e exógenos(1,6). É conhecido que os colangiócitos, além de produzirem e transportarem a bile, participam ativamente na reparação e remodelamento do epitélio biliar, em especial os pequenos colangiócitos, que mantêm características totipotentes e atuam na reparação do dano hepatocelular(1,5,6). De modo geral, o mecanismo fisiopatológico das colangiopatias envolve um insulto primário aos colangiócitos, o que faz com que alguns sofram apoptose, outros se tornem senescentes e outros, reativos(6). Os colangiócitos reativos secretam citocinas pró-inflamatórias, quimiostáticas e fatores de crescimento, que, por sua vez, recrutam células inflamatórias e mesenquimais e estimulam a angiogênese. O desequilíbrio entre essas respostas leva a fibrose periportal, ductopenia e, por fim, cirrose biliar(1,5,6). Os colangiócitos senescentes podem permanecer em um estado inerte ou tornarem-se hipersecretores de citocinas pró-inflamatórias, induzindo também resposta inflamatória e fibrose(6). Estes últimos possuem ainda maior suscetibilidade para malignização, estando mais relacionados ao desenvolvimento de colangiocarcinoma(6). Por se tratar de um grupo heterogêneo de doenças, uma recente classificação foi proposta de modo a agrupar as colangiopatias em diferentes categorias etiológicas: congênitas, imunomediadas, infecciosas, malignas, idiopáticas e outras (Tabela 1)(6). Discutiremos a seguir algumas causas não neoplásicas, incluindo a colangiopatia iatrogênica, e forneceremos um algoritmo diagnóstico útil na diferenciação entre elas, adotando a combinação de sinais observados na ultrassonografia (US), tomografia computadorizada (TC) e ressonância magnética (RM), com os seus respectivos aspectos epidemiológicos, clínicos e laboratoriais.  CONGÊNITAS Síndrome de Caroli Trata-se de uma doença autossômica recessiva rara, relacionada a malformação da placa ductal, com consequente processo inflamatório crônico e remodelamento. O acometimento dos grandes ductos biliares intra-hepáticos leva à doença de Caroli, caracterizada por dilatações fusiformes e saculares que se comunicam com o restante da via biliar. Entretanto, quando o comprometimento ocorre ao nível dos ductos interlobulares, o resultado é a fibrose hepática congênita. A síndrome de Caroli se desenvolve pela associação entre essas duas condições(7,8). Ela pode, ainda, associar-se a alterações renais, entre elas o rim espongiomedular e a doença renal policística autossômica dominante e recessiva(8). Os sintomas estão relacionados com as suas complicações, como hepatolitíase, colangite, abscesso hepático e insuficiência hepática(7,8). Os exames de imagem demonstram dilatações císticas das vias biliares intra-hepáticas (mais comumente no lobo hepático direito), estenoses biliares focais, hepatolitíase e fibrose. Em alguns casos, dilatações fusiformes das vias biliares extra-hepáticas podem também estar presentes como resultado da passagem de cálculos e colangites de repetição. Um achado típico da doença é o sinal do ponto central, que representa o feixe fibrovascular no interior ou na periferia da dilatação biliar (Figura 1)(7,8). A colangiopancreatografia por RM (CPRM) pode demonstrar características bastante sugestivas desta doença, como as citadas acima, e em casos duvidosos pode-se ainda complementar a investigação com a RM com contraste hepatoespecífico. Nesta modalidade diagnóstica é possível confirmar a comunicação dos cistos com a via biliar, uma vez que o contraste, excretado pelos hepatócitos, preenche a via biliar e os cistos em aquisições tardias, o que pode ocorrer entre 10 minutos e 24 horas (Figura 1)(9,10). 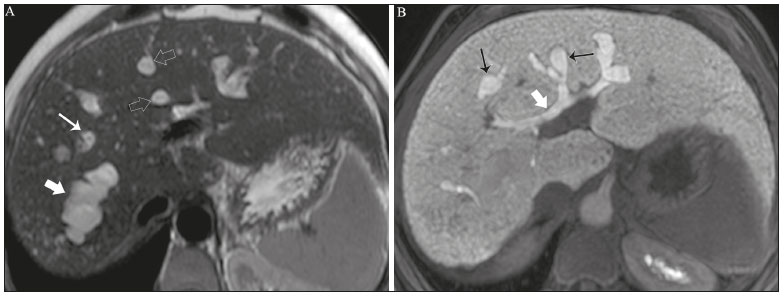 Figura 1. Em A (axial T2) e B (axial T1 pós-contraste hepatoespecífico tardio) há dilatação da via biliar intra-hepática (setas grossas), cistos esparsos (setas vazadas), alguns com ponto central de hipossinal em T2 (seta branca fina), com preenchimento pelo meio contraste na fase tardia (setas pretas), demonstrando haver comunicação com a via biliar, compatível com doença de Caroli. Dentre os seus principais diagnósticos diferenciais estão a colangite esclerosante primária e a colangite piogênica recorrente. Ambas se apresentam como dilatações biliares mais frequentemente fusiformes e esparsas, intercalando-se com estenoses segmentares, às vezes associadas a cálculos, sendo a colangite esclerosante primária frequentemente associada a doenças inflamatórias intestinais. Destaca-se que a presença de dilatações saculares é o principal aspecto a ser observado; se presente, favorece o diagnóstico de doença de Caroli(8). Fibrose hepática congênita A fibrose hepática congênita pertence ao grupo de doenças relacionadas a malformação da placa ductal. Possui herança autossômica recessiva e, assim como a doença de Caroli, apresenta associação com a doença renal policística autossômica dominante, doença renal policística autossômica recessiva e o rim espongiomedular(11,12). A doença acomete os ductos biliares interlobulares, e por isso não se manifesta macroscopicamente com dilatação das vias biliares, mas sim com fibrose hepática e hipertensão portal. Quando há dilatação das vias biliares concomitante, deve-se pensar em associação com outras entidades do grupo das malformações da placa ductal, como doença de Caroli, hamartomas biliares e cisto de colédoco(11). Por cursar de forma crônica e insidiosa, os sintomas surgem apenas na adolescência ou na fase adulta precoce, relacionados a complicações da hipertensão portal(11,12). Apesar de o diagnóstico definitivo ser histopatológico, a imagem tem conquistado crescente importância. Em cerca de 80% dos pacientes encontram-se pelo menos três dos seguintes achados: alterações morfológicas hepáticas (hipertrofia do segmento medial do lobo esquerdo, hipertrofia do lobo caudado e atrofia do lobo direito), esplenomegalia, circulação colateral portossistêmica, alterações renais e outras malformações da placa ductal, como hamartomas e cistos peribiliares (Figura 2)(12). 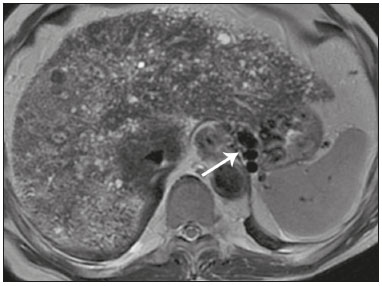 Figura 2. Inúmeros cistos hepáticos e sinais de hipertensão portal com circulação colateral perigástrica (seta). Paciente apresentava fibrose na elastografia por RM (valor de 5,9 kPa). Tais achados sugerem o diagnóstico de fibrose hepática congênita. Doença policística hepática A doença policística hepática tem herança autossômica dominante e faz parte do espectro de doenças congênitas de malformação da placa ductal, agora envolvendo os ductos biliares intra-hepáticos de médio calibre, e é por esta razão que esta entidade também é descrita no âmbito das colangiopatias. Durante a involução da placa ductal, os ductos biliares perdem a comunicação com a via biliar normal e dilatam-se progressivamente no decorrer da vida, sendo por isso conhecida como doença policística hepática do adulto(13,14). Dois espectros de doença são incluídos nesta categoria: doença hepática policística autossômica dominante com acometimento hepático isolado e doença renal policística autossômica dominante, forma mais comum com acometimento renal e hepático concomitante(1,14). Os pacientes são assintomáticos e apenas raramente apresentam sintomas relacionados a hepatomegalia, como dor, distensão abdominal e dispneia, ou secundários a rotura, sangramento ou infecção dos cistos. A evolução para insuficiência hepática é incomum(13,14). Os achados de imagem se resumem a um fígado de dimensões aumentadas à custa de inúmeros cistos periféricos ou peribiliares que não se comunicam com a árvore biliar, por vezes com sinais de sangramento ou calcificações periféricas(13). As dimensões e o número de cistos são variáveis, não existindo uma quantidade bem definida que diferencie entre a doença cística hereditária (doença policística hepática) da não hereditária(13,14). Fibrose cística O acometimento hepatobiliar acontece em até 40% dos pacientes com fibrose cística, sendo a cirrose hepática a segunda causa de morte nos pacientes portadores dessa doença(15). O conteúdo espesso e de composição anormal nas vias biliares leva a um processo inflamatório crônico, fibrose periportal e formação de cálculos biliares, que culminam com fibrose biliar focal, estenoses e dilatações das vias biliares intra-hepáticas e extra-hepáticas, bem como espessamento e atrofia da vesícula biliar, aspecto característico da doença. Em raros casos, pode haver progressão para fibrose multilobular e cirrose com hipertensão portal(16). Os exames de imagem podem revelar anormalidades das vias biliares, presentes em aproximadamente 50% dos pacientes mesmo sem doença hepática conhecida(15). Achado comum é a hiperecogenicidade periportal na US, que na RM se correlaciona mais frequentemente a deposição gordurosa do que a fibrose propriamente dita, havendo correlação positiva com estudos histopatológicos(16). Outro achado é a atrofia da vesícula biliar, que pode estar relacionada a estenose do ducto cístico e/ou a presença de muco espesso(15,16). Mediante a descrição dos achados, é possível perceber que essas manifestações hepatobiliares se sobrepõem às relacionadas à colangite esclerosante primária, seu principal diagnóstico diferencial(16). A distinção entre elas é feita pela associação com sinais típicos de fibrose cística em outros órgãos (por exemplo: lipossubstituição do parênquima pancreático, calcificações e cistos pancreáticos, esteatose e bronquiectasias) ou pela associação com doenças inflamatórias intestinais na colangite esclerosante primária (Figura 3)(1618). 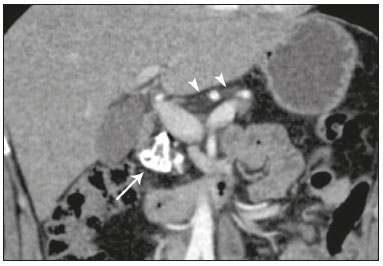 Figura 3. Paciente portador de fibrose cística, com pâncreas atrófico e lipossubstituído (cabeças de setas), apresentando calcificações grosseiras na sua porção cefálica (seta). IMUNOMEDIADAS Colangite biliar primária A colangite biliar primária, anteriormente chamada de cirrose biliar primária, é uma doença autoimune de causa ainda desconhecida que acomete principalmente mulheres entre a quinta e sétima décadas de vida, caracterizada por processo inflamatório crônico não supurativo dos ductos biliares interlobulares, resultando no desenvolvimento de fibrose(19,20). Nos estágios iniciais é mais frequentemente assintomática, podendo manifestar-se por fadiga e prurido intenso em até 50% dos casos e que pode anteceder a icterícia. Evolui naturalmente para cirrose, sendo o principal tratamento clínico efetivo o uso de ácido ursodesoxicólico, útil apenas nos estágios iniciais. Quando os pacientes evoluem para cirrose hepática, o único tratamento curativo é o transplante hepático(19). O diagnóstico é feito pela presença de dois entre três critérios: aumento de enzimas colestáticas por mais de seis meses, presença de anticorpos antimitocondriais e biópsia hepática compatível e caracterizada por colangite destrutiva não supurativa(19). Não é infrequente a associação com outras doenças autoimunes, como, por exemplo, a síndrome de Sjögren(21,22). Como a colangite biliar primária afeta os ductos biliares interlobulares estruturas microscópicas , os achados de imagem são sutis e muitas vezes tardios, surgindo após o estabelecimento da cirrose. Habitualmente, as vias biliares apresentam aspecto radiológico normal ou nota-se ductopenia nos exames de CPRM, denominado aspecto em árvore desfolhada (23). Mais recentemente, foi descrito o sinal do halo periportal na RM, que consiste na presença de um halo de hipossinal ao redor dos ramos portais periféricos, que se correlaciona com a fibrose periportal, mais bem caracterizado nas fases portal e de equilíbrio (Figura 4). Está intimamente relacionado com o estágio da doença, sendo mais exuberante nos estágios mais avançados(19). Linfonodomegalia regional também é achado frequente nos pacientes com colangite biliar primária, notadamente no hilo hepático. Outros achados são os relacionados a cirrose e hipertensão portal, inclusive com a presença de hiperplasia nodular regenerativa(19,24). 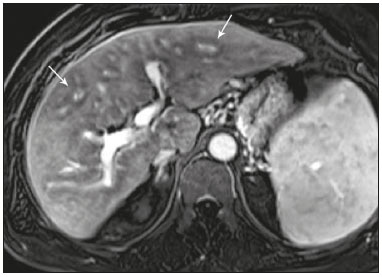 Figura 4. Halo de hipossinal periportal (setas) em paciente com colangite biliar primária. IDIOPÁTICAS Colangite esclerosante primária A colangite esclerosante primária é decorrente de processo inflamatório crônico idiopático das vias biliares intra-hepáticas e extra-hepáticas. Acredita-se que se trata de uma doença autoimune, principalmente pela forte associação com doenças inflamatórias intestinais, presente em 6080% dos casos, notadamente com a retocolite ulcerativa e em menor proporção com a doença de Crohn. Manifestase entre a quarta e quinta décadas de vida, com predileção para o sexo masculino. Os sintomas são inespecíficos e incluem síndrome colestática e dor abdominal. Frequentemente, os indícios iniciais resumem-se à elevação de enzimas colestáticas, como a fostatase alcalina e a gama-GT(25). O envolvimento crônico das vias biliares intra-hepáticas e extra-hepáticas leva a fibrose periductal progressiva, que pode evoluir para cirrose hepática. Essas características são comuns a muitas outras colangites esclerosantes. A colangite esclerosante primária não possui nenhum aspecto clínico, laboratorial, de imagem ou histopatológico específico, e seu diagnóstico é feito pela exclusão de causas secundárias como CE-IgG4, colangiopatia relacionada a síndrome da imunodeficiência adquirida, colangite eosinofílica, colangite isquêmica e colangite piogênica recorrente, descritas nas seções seguintes(17). O exame de escolha é a CPRM, que pode revelar acometimento multifocal, na maioria das vezes concomitante, dos ductos intra-hepáticos e extra-hepáticos, com segmentos curtos estreitados e irregulares intercalados com segmentos dilatados ou normais, configurando aspecto em colar de contas. Com a evolução da doença, os ductos periféricos tornam-se obliterados, não sendo mais caracterizados na RM, o que leva à impressão de árvore podada. Divertículos biliares podem estar presentes, e em menor porcentagem de pacientes (8%) é possível encontrar cálculos biliares (Figura 5)(17). O acometimento crônico causa importantes alterações morfológicas hepáticas, caracterizadas por atrofia dos segmentos laterais do lobo esquerdo e posteriores do lobo direito com hipertrofia compensatória do lobo caudado, e que podem estar associadas a macronódulos de regeneração com distribuição central(25). 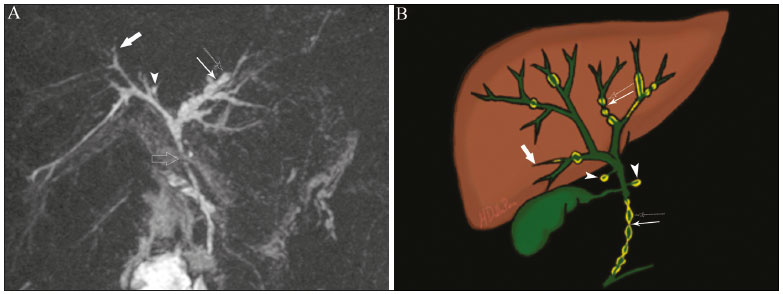 Figura 5. Paciente masculino de 38 anos com retocolite ulcerativa e colangites de repetição. Em A (CPRM) há estenose do colédoco (seta vazada) associada a pequena dilatação irregular das vias biliares intra-hepáticas com áreas de estenose (seta fina contínua) intercaladas com segmentos dilatados (seta tracejada), com aspecto em colar de contas; divertículos biliares (cabeça de seta); e obliteração dos ductos biliares periféricos configurando um aspecto de árvore podada (seta grossa). Os achados de imagem e o contexto clínico do paciente sugerem o diagnóstico de colangite esclerosante primária. Representação gráfica em B. Pacientes com colangite esclerosante primária têm entre 1015% de chance de desenvolver colangiocarcinoma, sendo esta uma das mais temidas complicações, ocorrendo mais comumente quando o paciente apresenta associação de colangite esclerosante primária com doenças inflamatórias intestinais. Uma dificuldade que se interpõe é que o colangiocarcinoma inicial divide características comuns com a colangite esclerosante primária, como estreitamento luminal e espessamento parietal, por exemplo, o que limita a sua identificação e eventualmente torna essas duas entidades indistinguíveis(17). Para essa distinção, deve-se considerar a extensão do segmento estenótico, a espessura, o hiper-realce e a assimetria parietal, bem como o grau de irregularidade luminal e o nível sérico de CA 19-9, este geralmente bastante elevado no colangiocarcinoma(2628). É importante salientar que não há nenhum tratamento efetivo para colangite esclerosante primária, sendo o transplante hepático a única terapêutica curativa e com recorrência de até 25% em 510 anos após o transplante(17). Colangiopatia esclerosante relacionada à IgG4 A colangiopatia relacionada à IgG4 (CE-IgG4) referese ao acometimento biliar da doença esclerosante relacionada à IgG4 que, por ser sistêmica, pode se manifestar em múltiplos órgãos. O sítio biliar é o segundo em frequência de acometimento, atrás apenas do sítio pancreático, embora ambos estejam concomitantemente envolvidos na maioria dos casos(29). Apenas cerca de 10% dos pacientes com CEIgG4 não apresentam pancreatite autoimune(30). Outras doenças autoimunes frequentemente associadas são: nefrite, fibrose retroperitonial, mesenterite esclerosante e sialoadenite(29). Os pacientes acometidos são no geral homens com idade média de 60 anos. Clinicamente, apresentam síndrome colestática e sintomas relacionados a pancreatite autoimune, como perda de peso, dor abdominal e diabetes(29). Os exames laboratoriais demonstram altos níveis de IgG4 sérica, marcador bastante específico, além de positividade para fator reumatoide e anticorpo antinuclear(30). Em alguns casos, a doença é autolimitada, enquanto em outros evoluem para cirrose. Uma característica marcante da doença é a excelente resposta a corticoterapia(29). A CE-IgG4 é doença de ductos biliares intra-hepáticos e extra-hepáticos, sendo estes últimos os mais frequentemente envolvidos, o que ocorre isoladamente em 43% dos pacientes. Na TC e RM, os ductos acometidos apresentam espessamento, irregularidade e realce pelo meio de contraste, de forma focal ou difusa. A presença de dilatação pré-estenótica é característica marcante da doença. Os segmentos são geralmente longos e contínuos (Figura 6), sendo às vezes possível a identificação de um tecido que os envolve circunferencialmente. Além disso, pode haver espessamento parietal da vesícula biliar, com hipoecogenicidade na US e hipointensidade de sinal na RM(29).  Figura 6. Na CPRM (A) há segmento longo de estenose do colédoco (seta), com dilatação da via biliar a montante. Em B (axial T1 pré-contraste) o pâncreas apresenta redução difusa do sinal e perda das lobulações (asteriscos), achados sugestivos de pancreatite autoimune. O contexto clínico e os achados de imagem sugerem colangiopatia relacionada a IgG4. Representação gráfica em C adicionalmente demonstrando dilatação pré-estenótica (cabeças de setas) das vias biliares intra-hepáticas. Tais aspectos fazem da CE-IgG4 o grande mimetizador do colangiocarcinoma e da colangite esclerosante primária. O diferencial pode ser feito pela presença de comprometimento de outros órgãos, como pâncreas e rim, e pelos altos níveis séricos de IgG4 (≥ 300 mg/dL). Diante da suspeita de colangite esclerosante primária, outras características podem ser úteis, entre elas idade do paciente (geralmente mais jovens, abaixo dos 40 anos), associação com doenças inflamatórias intestinais, envolvimento biliar multifocal com segmentos estreitados seguidos de áreas normais ou dilatadas, além da presença de divertículos biliares(29,30). Ainda assim, a diferenciação com essas doenças pode não ser possível, e nessa situação o estudo histopatológico torna-se necessário(30). Colangite eosinofílica A colangite eosinofílica é doença rara de causa desconhecida, com alguns poucos casos descritos na literatura. Caracteriza-se por infiltração de eosinófilos no sistema biliar e fibrose(31). Esta entidade associa-se a outras doenças também caracterizadas por infiltração eosinofílica, como a gastroenterite eosinofílica, e que apresentam como característica marcante a eosinofilia periférica, presente em aproximadamente 50% dos pacientes. Geralmente, os pacientes apresentam boa resposta com corticoterapia(31). Os achados de imagem são inespecíficos, podendo haver acometimento focal ou difuso das vias biliares intrahepáticas e extra-hepáticas, com espessamento parietal e estreitamento luminal, associado a espessamento parietal da vesícula biliar, este último um sinal que pode ajudar na suspeita radiológica dessa doença (Figura 7)(17). 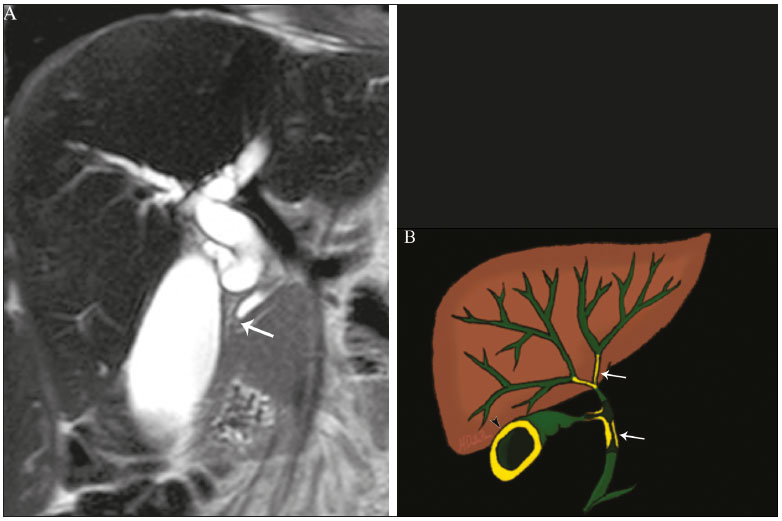 Figura 7. Homem de 36 anos com antecedente de pneumonia eosinofílica apresentando dor abdominal, icterícia e eosinofilia periférica. Em A (coronal T2) há afilamento abrupto do colédoco distal intrapancreático (seta), com dilatação das vias biliares a montante. O estudo anatomopatológico foi compatível com colangite eosinofílica. Representação gráfica em B também demonstrando espessamento parietal da vesícula biliar (cabeça de seta). INFECCIOSAS Colangite piogênica recorrente A colangite piogênica recorrente é frequente na Ásia, onde há endemia de infecção por Clonorchis sinensis, parasita associado à doença(32). Outras parasitoses relacionadas a colangite piogênica recorrente são a ascaridíase e a fasciolose. Acredita-se que a infestação por esses parasitas pode lesar o epitélio ductal, preferencialmente intrahepático, causando dilatação biliar sem fator obstrutivo(25). Isto pode acarretar estase de bile, estreitamentos luminais e hepatolitíase, que também pode ser decorrente de fatores dietéticos e de outros agentes, como Escherichia coli, Schistosoma mansoni e Helicobacter pylori. Alterações estruturais das vias biliares, tais como doença de Caroli, colangite isquêmica e colangite esclerosante primária, são fatores predisponentes à ocorrência de colangite piogênica recorrente. Os sintomas estão relacionados a colangites agudas recorrentes, lembrando que a doença ocasionalmente pode ser assintomática, cursando apenas com alterações laboratoriais, como elevação de enzimas colestáticas(17). Os cálculos pigmentados que se formam nas vias biliares intra-hepáticas podem ou não surgir junto a um estreitamento ductal e são hiperdensos na TC, isointensos em T2 e hiperintensos em T1 na RM. Há também atrofia do parênquima no segmento acometido, ocorrendo preferencialmente nos segmentos laterais do lobo esquerdo e posteriores do lobo direito, com hipertrofia dos demais(25). O risco de desenvolver colangiocarcinoma também está aumentado nos pacientes com colangite piogênica recorrente, ocorrendo em até 5% deles. A neoplasia acomete preferencialmente os segmentos atrofiados ou os segmentos com múltiplos cálculos(32). Seu principal diagnóstico diferencial é a colangite esclerosante primária, distinguindo-se desta por não apresentar aspecto em contas ou divertículos biliares, pelo fato de seu acometimento ser preferencialmente intrahepático isolado, e por apresentar uma desproporção da dilatação da via biliar, com distribuição preferencialmente periférica(17). VASCULAR Colangiopatia isquêmica Como já mencionado, os ductos biliares são supridos pelo plexo peribiliar, formado por ramos da artéria hepática, e por isso suscetíveis a redução no suprimento vascular arterial. Os segmentos mais acometidos são o terço médio do colédoco e a confluência dos ductos hepáticos, que podem ser afetados de forma focal ou multifocal(33). A lesão isquêmica da via biliar pode ser dividida, grosseiramente, em três tipos: necrose com formação de bilomas, descamação epitelial com obstrução biliar e fibrose com estenose ductal(17). O primeiro ocorre geralmente como consequência de insuficiência arterial severa com necrose de toda a espessura da parede do ducto, seguida de descontinuidade parietal e formação de bilomas (Figura 8). Estes são representados em exames de imagem como coleções líquidas contíguas aos ductos lesados(33). O segundo é decorrente de um suprimento arterial insuficiente mais brando, com descamação do epitélio, que se acumula no lúmen do ducto, podendo levar a obstrução biliar. Esse epitélio descamado forma moldes intraductais, que na RM são hiperintensos em T1 e hipointensos em T2, semelhantes a cálculos (Figura 8)(25,33). Cronicamente, esses dois tipos de acometimento ocasionam fibrose e estenose ductal, que pode ser focal ou difusa. Nesta fase, os achados de colangiopatia isquêmica podem se assemelhar à colangite esclerosante primária. Portanto, o contexto clínico adequado e a evidência direta de trombose da artéria hepática são fundamentais para o raciocínio diagnóstico(17). 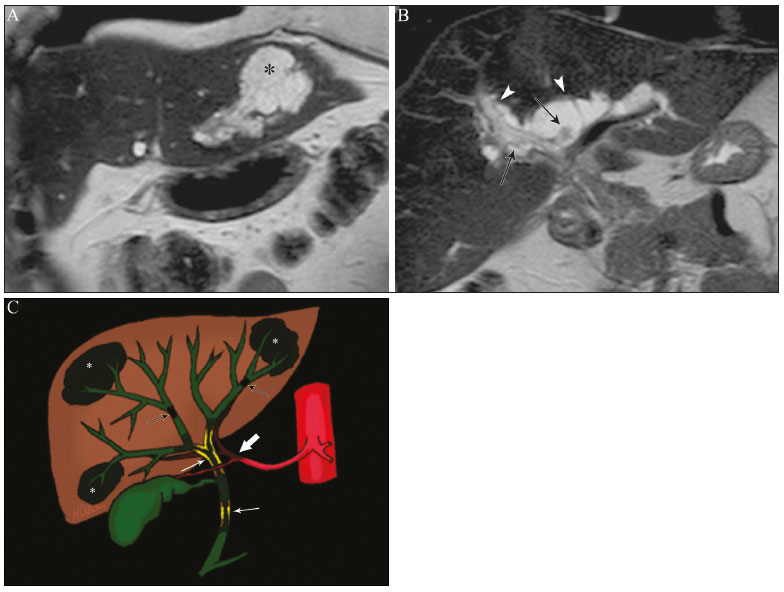 Figura 8. Paciente com trombose da artéria hepática após dois anos de transplante hepático. Em A e B (coronal T2) há acentuada dilatação das vias biliares (cabeças de setas), conteúdo hipointenso intraductal (setas pretas) e um biloma (asterisco) contíguo à via biliar dilatada. O contexto clínico e o conjunto dos achados de imagem sugerem colangiopatia isquêmica. A representação gráfica em C demonstra, além desses achados, estenose do ducto hepático (setas brancas finas) e trombose da artéria hepática (seta grossa). Numerosas causas provocam obstrução de artérias e arteríolas hepáticas. Entre as mais comumente associadas a colangiopatia isquêmica podemos citar a trombose póstransplante hepático, a quimioembolização intra-arterial, a lesão vascular iatrogênica após cirurgias do trato gastrointestinal, a telangiectasia hemorrágica hereditária e a poliarterite nodosa(33,34). A trombose arterial é a complicação vascular mais comum pós-transplante e ocorre geralmente entre 15 dias e 6 meses após o procedimento, sendo a segunda causa mais comum de perda do enxerto, após a rejeição(35). IATROGÊNICA A lesão iatrogênica da via biliar é responsável por 95% das estenoses benignas da via biliar. Em decorrência da sua alta prevalência, deve ser prontamente reconhecida, tendo os métodos de imagem um papel central no seu diagnóstico. Cerca de 80% dos casos são diagnosticados dentro de um ano da cirurgia, podendo evoluir para cirrose hepática se não tratados adequadamente(36). Os procedimentos cirúrgicos que oferecem risco de lesão iatrogênica da via biliar incluem os que envolvem manipulação do ligamento hepatoduodenal, sendo a colecistectomia laparoscópica a mais frequentemente associada(36). As causas são diversas, sendo a principal delas a identificação incorreta do colédoco(36), o que enfatiza a importância da CPRM pré-operatória, que pode identificar eventuais variações anatômicas biliares. Outra importante ferramenta mais recentemente disponível é a RM com contraste hepatoespecífico, que fornece informações funcionais da via biliar, contribuindo para uma maior acurácia na identificação de variações anatômicas, principalmente nos pacientes sem dilatação biliar, e ainda na caracterização de estenoses e fístulas biliares pós-operatórias(10,37). As variações anatômicas mais frequentes são: ducto colédoco cursando paralelo a ducto aberrante; ducto cístico longo, com inserção medial baixa; ducto aberrante direito drenando para a via biliar esquerda; e confluência tripla da via biliar intra-hepática (Figura 9)(38). 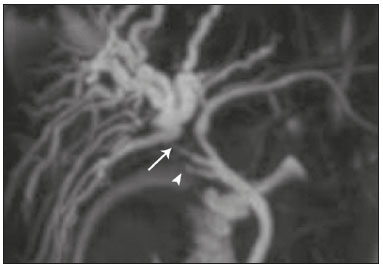 Figura 9. Paciente em pós-operatório de colecistectomia com estenose iatrogênica do ducto hepático direito (seta), que apresenta drenagem baixa no ducto hepático comum, na proximidade do coto cístico (cabeça de seta). O tratamento inicial de escolha é a dilatação endoscópica ou percutânea da via biliar, sendo a reconstrução cirúrgica reservada aos casos de insucesso terapêutico(36). ALGORITMO DIAGNÓSTICO Antes de estreitarmos o diagnóstico das causas benignas de colangiopatias, precisamos excluir a possibilidade de colangiocarcinoma. Para isso, uma série de aspectos de imagem que favoreçam malignidade deve ser analisada, dentre eles: segmento estenótico longo (> 12 mm); espessura da parede do ducto (> 3,0 mm); hiper-realce em relação ao parênquima hepático na fase portal ou de equilíbrio; assimetria parietal; e irregularidade luminal(26,28). Isoladamente, esses achados têm baixa acurácia diagnóstica, mas quando analisados em conjunto, a acurácia pode chegar a 90%(26,28). A dosagem sérica de marcadores tumorais também contribui para uma maior suspeição de neoplasia, sendo o CA 19-9 o mais amplamente utilizado(39). Trata-se de um marcador tumoral expresso pelo epitélio pancreatobiliar, tanto em condições benignas como malignas, e por vários tumores epiteliais de outros sítios(40,41). Em pacientes com colestase sem antecedente de colangite esclerosante primária, o ponto de corte proposto é 90 U/mL (sensibilidade de 61% e especificidade de 95%)(40); em pacientes com antecedente de colangite esclerosante primária este valor passa a ser 129 U/mL (sensibilidade de 78,6% e especificidade de 98,5%). Neste sentido, é interessante observar que até 33% dos pacientes com colangite esclerosante primária apresentam elevação do CA 19-9 acima desse valor de corte, podendo se encontrar muito aumentado(27). Outras causas de elevação do CA 19-9 são a cirrose e a pancreatite aguda e crônica(40). Se afastada a possibilidade de colangiocarcinoma, devemos considerar não só o aspecto de imagem das lesões, mas também aspectos epidemiológicos e laboratoriais. Com essa finalidade, propusemos uma abordagem algorítmica que irá auxiliar o diagnóstico, reduzindo o tempo entre início do quadro clínico e introdução do tratamento adequado (Figura 10). 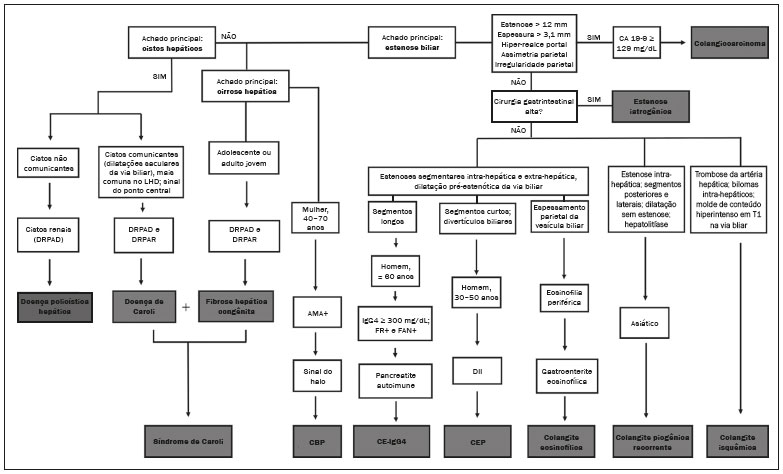 Figura 10. Algoritmo diagnóstico das colangiopatias não neoplásicas. LHD, lobo hepático direito; DRPAD, doença renal policística autossômica dominante; DRPAR, doença renal policística autossômica recessiva; AMA, anticorpo antimitocondrial; FR, fator reumatoide; FAN, fator antinuclear; CE-IgG4, colangite esclerosante por IgG4; DII, doença inflamatória intestinal; CBP, colangite biliar primária; CEP, colangite esclerosante primária. CONCLUSÃO O aspecto de imagem, bem como os sinais e sintomas das colangiopatias não tumorais são semelhantes entre si e com os observados em outros contextos, como, por exemplo, na colangiopatia pós-traumática(42) ou mesmo na de origem tumoral, quando eventualmente será necessária a coleta de material para análise histológica(43). Apenas uma análise conjunta de todos os aspectos radiológicos, clínicos e epidemiológicos de cada uma dessas entidades é capaz de estreitar as possibilidades diagnósticas, contribuindo para a adoção de terapêuticas precoces. REFERÊNCIAS 1. Lazaridis KN, LaRusso NF. The cholangiopathies. Mayo Clin Proc. 2015;90:791800. 2. U.S. Department on Health and Human Services. Organ Procurement and Transplantation Network. National Data [Internet]. [cited 2018 Jun 16]. Available from: https://optn.transplant.hrsa.gov/data/ view-data-reports/national-data/ 3. Menias CO, Surabhi VR, Prasad SR, et al. Mimics of cholangiocarcinoma: spectrum of disease. Radiographics. 2008;28:111529. 4. Walshe TM, McLean KA, Patel R, et al. Imaging of the biliary tree: infection, inflammation and infiltration. Appl Radiol. 2016;45:206. 5. OHara SP, Tabibian JH, Splinter PL, et al. The dynamic biliary epithelia: molecules, pathways, and disease. J Hepatol. 2013;58:575 82. 6. Cheung AC, Pisarello MJL, LaRusso NF. Pathobiology of biliary epithelia. Biochim Biophys Acta Mol Basis Dis. 2018;1864(4 Pt B): 122031. 7. Levy AD, Rohrmann CA. Diseases of the gallbladder and bile ducts. In: Gourtsoyiannis NC, Ros PR, editors. Radiologic-pathologic correlations from head to toe. 1st ed. Berlin Heidelberg: Springer; 2005. p. 50932. 8. Levy AD, Rohrmann CA Jr, Murakata LA, et al. Carolis disease: radiologic spectrum with pathologic correlation. AJR Am J Roentgenol. 2002;179:10537. 9. Salvadori PS, Torres US, DIppolito G. Contrast-enhanced magnetic resonance cholangiography with gadoxetic-acid-disodium for the detection of biliary-cyst communication in Caroli disease. Gastroenterol Hepatol. 2015;39:66970. 10. Lee NK, Kim S, Lee JW, et al. Biliary MR imaging with Gd-EOBDTPA and its clinical applications. Radiographics. 2009;29:1707 24. 11. Akhan O, Karaosmanoglu AD, Ergen B. Imaging findings in congenital hepatic fibrosis. Eur J Radiol. 2007;61:1824. 12. Zeitoun D, Brancatelli G, Colombat M, et al. Congenital hepatic fibrosis: CT findings in 18 adults. Radiology. 2004;231:10916. 13. Brancatelli G, Federle MP, Vilgrain V, et al. Fibropolycystic liver disease: CT and MR imaging findings. Radiographics. 2005;25:659 70. 14. Morgan DE, Lockhart ME, Canon CL, et al. Polycystic liver disease: multimodality imaging for complications and transplant evaluation. Radiographics. 2006;26:165568. 15. Robertson MB, Choe KA, Joseph PM. Review of the abdominal manifestations of cystic fibrosis in the adult patient. Radiographics. 2006;26:67990. 16. King LJ, Scurr ED, Murugan N, et al. Hepatobiliary and pancreatic manifestations of cystic fibrosis: MR imaging appearances. Radiographics. 2000;20:76777. 17. Seo N, Kim SY, Lee SS, et al. Sclerosing cholangitis: clinicopathologic features, imaging spectrum, and systemic approach to differential diagnosis. Korean J Radiol. 2016;17:2538. 18. Lavelle LP, McEvoy SH, Mhurchu EN, et al. Cystic fibrosis below the diaphragm: abdominal findings in adult patients. Radiographics. 2015;35:68095. 19. Meng Y, Liang Y, Liu M. The value of MRI in the diagnosis of primary biliary cirrhosis and assessment of liver fibrosis. PLoS One. 2015;10:e0120110. 20. Beuers U, Gershwin ME, Gish RG, et al. Changing nomenclature for PBC: from cirrhosis to cholangitis. Gastroenterology. 2015;149: 16279. 21. Lins L, Paraná R, Reis SRA, et al. Primary biliary cirrhosis and primary Sjögrens syndrome: insights for the stomatologist. Case Rep Gastroenterol. 2014;8:2516. 22. Selmi C, Meroni PL, Gershwin ME. Primary biliary cirrhosis and Sjogrens syndrome: autoimmune epithelitis. J Autoimmun. 2012; 39:3442. 23. Yeh MJ, Kim SY, Jhaveri KS, et al. Imaging of autoimmune biliary disease. Abdom Radiol. 2017;42:318. 24. Wenzel JS, Donohoe A, Ford KL 3rd, et al. Primary biliary cirrhosis: MR imaging findings and description of MR imaging periportal halo sign. AJR Am J Roentgenol. 2001;176:8859. 25. Leake R, Rezvani M, Willmore R, et al. Primary sclerosing cholangitis and its mimickers: a review of disease and MRI findings. Curr Radiol Rep. 2017;5:21. 26. Kim JY, Lee JM, Han JK, et al. Contrast-enhanced MRI combined with MR cholangiopancreatography for the evaluation of patients with biliary strictures: differentiation of malignant from benign bile duct strictures. J Magn Reson Imaging. 2007;26:30412. 27. Levy C, Lymp J, Angulo P, et al. The value of serum CA 19-9 in predicting cholangiocarcinomas in patients with primary sclerosing cholangitis. Dig Dis Sci. 2005;50:173440. 28. Yu XR, Huang WY, Zhang BY, et al. Differentiation of infiltrative cholangiocarcinoma from benign common bile duct stricture using three-dimensional dynamic contrast-enhanced MRI with MRCP. Clin Radiol. 2014;69:56773. 29. Vlachou PA, Khalili K, Jang HJ, et al. IgG4-related sclerosing disease: autoimmune pancreatitis and extrapancreatic manifestations. Radiographics. 2011;31:1379402. 30. Zen Y, Nakanuma Y. IgG4 cholangiopathy. Int J Hepatol. 2012; 2012:472376. 31. Nashed C, Sakpal SV, Shusharina V, et al. Eosinophilic cholangitis and cholangiopathy: a sheep in wolves clothing. HPB Surg. 2010;2010:906496. 32. Heffernan EJ, Geoghegan T, Munk PL, et al. Recurrent pyogenic cholangitis: from imaging to intervention. AJR Am J Roentgenol. 2009;192:W2835. 33. Deltenre P, Valla DC. Ischemic cholangiopathy. Semin Liver Dis. 2008;28:23546. 34. Wang Z, Wang M, Duan F, et al. Bile duct injury after transcatheter arterial chemoembolization: risk factors and clinical implications. Hepatogastroenterology. 2014;61:94753. 35. Singh AK, Nachiappan AC, Verma HA, et al. Postoperative imaging in liver transplantation: what radiologists should know. Radiographics. 2010;30:33951. 36. Jablonska B, Lampe P. Iatrogenic bile duct injuries: etiology, diagnosis and management. World J Gastroenterol. 2009;15:4097104. 37. Zattar-Ramos LC, Bezerra ROF, Siqueira LTDB, et al. Hepatocytespecific contrast agent-enhanced magnetic resonance cholangiography: perioperative evaluation of the biliary tree. Radiol Bras. 2017;50:38994. 38. Mortelé KJ, Ros PR. Anatomic variants of the biliary tree: MR cholangiographic findings and clinical applications. AJR Am J Roentgenol. 2001;177:38994. 39. Lee JJ, Schindera ST, Jang HJ, et al. Cholangiocarcinoma and its mimickers in primary sclerosing cholangitis. Abdom Radiol. 2017; 42:2898908. 40. Marrelli D, Caruso S, Pedrazzani C, et al. CA19-9 serum levels in obstructive jaundice: clinical value in benign and malignant conditions. Am J Surg. 2009;198:3339. 41. Venkatesh PGK, Navaneethan U, Shen B, et al. Increased serum levels of carbohydrate antigen 19-9 and outcomes in primary sclerosing cholangitis patients without cholangiocarcinoma. Dig Dis Sci. 2013;58:8507. 42. Muniz BC, Ribeiro BNF, Ventura N, et al. Hemobilia in a patient with arteriobiliary fistula after liver contusion. Radiol Bras. 2018; 51:4134. 43. Nunes TF, Tibana TK, Santos RFT, et al. Percutaneous transhepatic cholangiobiopsy. Radiol Bras. 2019;52:412. 11 Departamento de Diagnóstico por Imagem Escola Paulista de Medicina da Universidade Federal de São Paulo (EPM-Unifesp), São Paulo, SP, Brasil. Correspondência: Dra. Marina Silva Zacarias EPM-Unifesp Departamento de Diagnóstico por Imagem Rua Napoleão de Barros, 800, Vila Clementino São Paulo, SP, Brasil, 04023-062 E-mail: marina_sz@hotmail.com a. https://orcid.org/0000-0001-8553-974X b. https://orcid.org/0000-0002-6556-7003 c. https://orcid.org/0000-0003-0838-3731 d. https://orcid.org/0000-0002-8601-4774 e. https://orcid.org/0000-0002-2019-7918 f. https://orcid.org/0000-0002-2701-1928 Recebido para publicação em 31/5/2019 Aceito, após revisão, em 31/7/2019 Publicado em: 15/07/2020 |
|
Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554