Radiologia Brasileira - Publicação Científica Oficial do Colégio Brasileiro de Radiologia
AMB - Associação Médica Brasileira CNA - Comissão Nacional de Acreditação
 Vol. 49 nº 5 - Sep. / Oct. of 2016
Vol. 49 nº 5 - Sep. / Oct. of 2016
|
REVIEW ARTICLE
|
|
Tomography patterns of lung disease in systemic sclerosis |
|
|
Autho(rs): Andréa de Lima Bastos1; Ricardo de Amorim Corrêa2; Gilda Aparecida Ferreira3 |
|
|
Keywords: Sclerosis; Scleroderma, systemic; Radiology; Tomography, X-ray computed; Lung diseases. |
|
|
Abstract: INTRODUCTION
Systemic sclerosis (SSc) is an autoimmune connective tissue disease, of unknown cause, characterized by inflammatory changes, fibrosis, obstructive small-vessel vasculopathy, and collagen deposition in various organs, especially the lungs(1,2). The disease is more common in women than in men, at an average ratio ranging from 3:1 to 8:1, and its incidence peaks between 45 and 64 years of age(3,4). Pulmonary involvement is a major feature of the evolution of SSc, the incidence of such involvement ranging from 70% to 90%, and is currently the leading cause of morbidity and mortality among individuals with SSc(4). In SSc patients, inflammation and interstitial fibrosis are seen in SSc patients, as are excessive deposition of extracellular matrix and vascular obliteration, which can also cause pulmonary hypertension(5). The resulting anatomical changes have variable radiological manifestations. The early detection and proper interpretation of the radiological findings constitute a critical step in deciding when is the proper time to start treatment. The pulmonary aspect of the disease has a clinical course that can range from presentations that are more indolent to those that progress rapidly, with a corresponding rapid decline in lung function. In some cases, the pulmonary fibrosis precedes the appearance of systemic disease by a number of years. Respiratory symptoms, present in more than 50% of patients with SSc, do not constitute reliable indicators of impairment of the lung parenchyma, because they can also arise from the pulmonary vascular impairment or muscle weakness associated with the disease(2,6). The pulmonary changes arising from SSc can be demonstrated by diagnostic imaging methods. Such methods include high-resolution computed tomography (HRCT) of the chest, which allows the anatomy of the lung parenchyma to be detailed with a sensitivity far superior to that of X-ray, constituting an important tool for the diagnosis and evaluation of the extent of the disease(7). The aim of this study was to review the literature on the main pulmonary changes resulting from SSc and the corresponding radiological manifestations. METHODS We selected articles by conducting searches in the Medline (PubMed), Lilacs, and SciELO databases, using the following keywords: scleroderma; systemic sclerosis, radiology, computed tomography, and lung diseases. We further limited our searches to articles published in English and involving human subjects. Considering the approaches taken by the authors of studies employing HRCT and, primarily, changes in the consensus for interstitial lung disease, we selected 21 articles, published between 2000 and 2015, that included descriptive information related to radiological aspects of lung changes secondary to SSc. To identify additional relevant references, we performed hand searches of the bibliographies of the selected articles. DISCUSSION Interstitial changes There are various forms of pulmonary involvement in SSc, interstitial disease being the most common. Interstitial changes in SSc ares characterized by diffuse pulmonary distribution and can correspond to the various histopathological subtypes. The use of HRCT plays an important role in the topographic identification of SSc lesions, as well as in evaluating their extent and monitoring the evolution of the process. Initial descriptions of the pathological features of the disease come from post-mortem studies, which have suggested a pattern of fibrosis with little inflammatory response, although the presence of inflammation and fibrosis in the alveolar walls has recently been discussed(8). In one autopsy study, fibrosis was observed in more than 75% of the cases(9). Fibrosis caused by SSc is the most frequently observed histological component, with a variable clinical evolution(10). Of all cases of pulmonary fibrosis in SSc, 50.077.5% are due to nonspecific interstitial pneumonia, which could explain that difference in prognosis(10,11). Nonspecific interstitial pneumonia is a particular form of idiopathic interstitial pneumonia characterized by varying degrees of inflammation and fibrosis, from forms predominated by inflammatory processes to those predominated by fibrosis, which are the most common, although without the fibroblastic foci and honeycombing seen in usual interstitial pneumonia(8,12). The abnormalities seen in nonspecific interstitial pneumonia, most often observed in HRCT, include irregular, reticular, ground-glass attenuation, as well as traction bronchiectasis and bronchiolectasis (Figure 1). These lesions rarely occur in the subpleural regions of the lungs (Figure 2), and that aspect can be useful in differentiating nonspecific interstitial pneumonia from usual interstitial pneumonia(12). 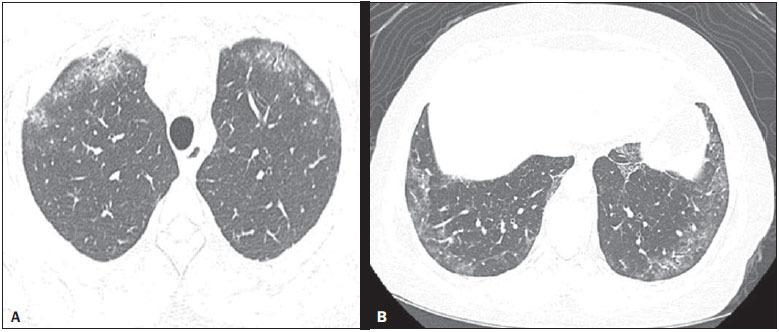 Figure 1. A 55-year-old female with an 8-year history of SSc, without respiratory symptoms. Note the bilateral areas of ground-glass attenuation in the cortical regions of the upper lobes (A) and of the basal segments of the lower lobes (B). 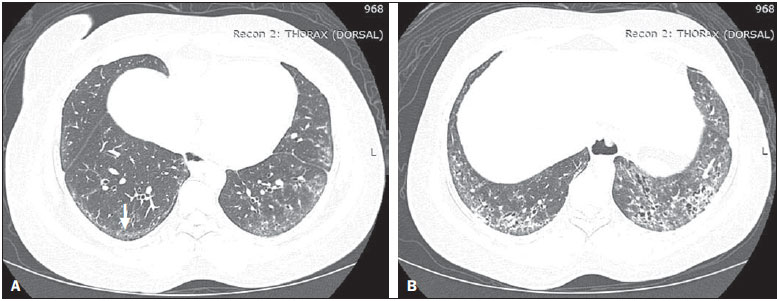 Figure 2. A 37-year-old female with a 10-year history of SSc, presenting with severe dyspnea and esophageal dilation. HRCT showing, in A, ground-glass attenuation, sparing the subpleural regions (arrow), and, in B, traction bronchiectasis, thickening of the interlobular septa, and subpleural lines, accompanied by ground-glass attenuation with a cortical distribution. Usual interstitial pneumonia is a less common presentation in SSc. The typical HRCT pattern in usual interstitial pneumonia is characterized by reticular changes, predominantly in the basal, peripheral, and subpleural regions, together with honeycombing, traction bronchiectasis, traction bronchiolectasis, architectural distortion, and volume loss in the affected region(13). This pattern has a worse prognosis than does that observed in nonspecific interstitial pneumonia (Figure 3). 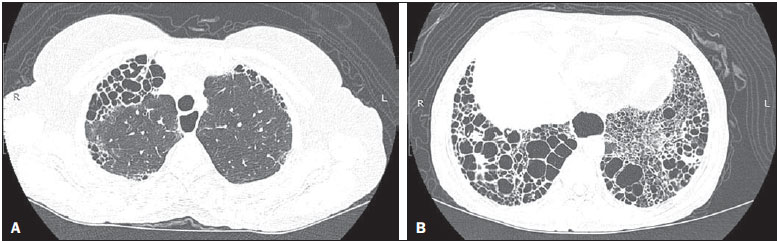 Figure 3. A 58-year-old female with a 26-year history of SSc, presenting with cough, pronounced dyspnea, and bronchospasm. HRCT showing significant esophageal dilation and extensive bilateral honeycombing, predominantly in the upper lobes (A) and lower lobes (B), with a pattern characteristic of usual interstitial pneumonia. Alveolar changes Less common forms of pulmonary involvement reported in SSc are diffuse alveolar damage, pulmonary hemorrhage, organizing pneumonia, aspiration pneumonia, and lung disease associated with the drugs used in the treatment(6). There have been only a few reported cases of organizing pneumonia as a manifestation of SSc. However, because the gold standard for diagnosis is open-lung biopsy, which is rarely performed in these patients, the true incidence of organizing pneumonia in SSc is unknown. Taylor et al.(14)reported three cases of organizing pneumonia diagnosed by open-lung biopsy in patients with SSc. HRCT can reveal pronounced bilateral consolidation in the subpleural space (Figure 4) or in the peribronchial areas, with a migratory aspect and some areas of ground-glass attenuation. The reversed halo sign can be useful in suggesting the diagnosis. Histologically, organizing pneumonia involves the alveoli and alveolar ducts, with or without intraluminal bronchial polyps(12). Clinically, the disease manifests as cough, fever, and dyspnea; there is a predominance of lymphocytes in the bronchoalveolar lavage fluid(12,14). Other authors have reported this same presentation(1517). 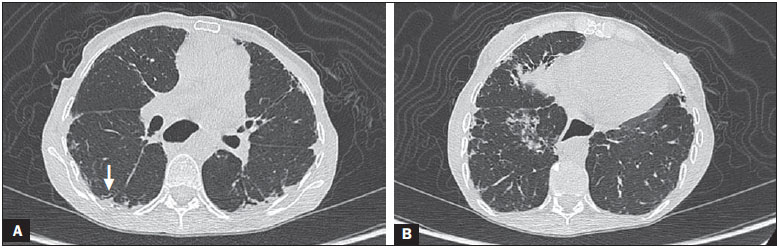 Figure 4. A 49-year-old female with a 10-year history of SSc, without respiratory symptoms. HRCT with a lung window, showing small subpleural focal consolidations interspersed with bronchiectasis (A, arrow) in the middle and lower thirds of the lung segments (A and B, respectively), together with marked esophageal dilation. The radiological pattern is suggestive of organizing pneumonia, even before the histopathological study. In the case of atypical clinical and radiological presentations, open-lung biopsy should be considered, given that organizing pneumonia and fibrosing alveolitis differ in terms of prognosis and treatment. The former responds to corticosteroids, whereas the latter often requires the use of immunosuppressive agents(14). Because pulmonary changes that are detectable by routine chest X-ray in only 2553% of cases can be detected with HRCT in up to 94% of cases, HRCT is the method of choice for the investigation of interstitial lung disease in SSc(18). In patients with SSc, aspiration pneumonia occurs due to esophageal involvement secondary to the disease. Esophageal involvement is seen in 5090% of cases of SSc, symptoms arising from dysmotility and reflux(19). On CT scans of SSc patients, the coronal diameter of the esophageal lumen is increased, ranging from 1.2 cm to 4.0 cm, with a mean of 2.3 cm(20). Vascular changes The pulmonary vascular involvement in SSc can cause pulmonary arterial hypertension, which results from the increase in pulmonary vascular resistance caused by occlusion and remodeling of the pulmonary arterioles. Pulmonary arterial hypertension, defined as a mean pulmonary artery pressure equal to or greater than 25 mmHg and a pulmonary artery occlusion pressure equal to or less than 15 mmHg, is a major cause of SSc-related morbidity and mortality (Figure 5), mean survival after diagnosis ranging from 1.5 to 3 years(21). 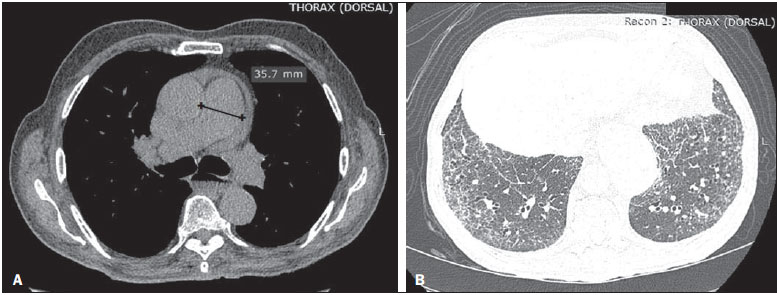 Figure 5. An 81-year-old male with a 10-year history of SSc. A: HRCT with a mediastinal window showing an increase in the caliber of the pulmonary artery trunk (35.7 mm) and esophageal dilation with an air-fluid level inside. B: Areas of ground-glass attenuation in the pulmonary parenchyma, with thickening of the interlobular septa, interspersed with traction bronchiectasis, predominantly cortical in distribution, suggestive of nonspecific interstitial pneumonia. This manifestation can occur in isolation or can be accompanied by interstitial lung disease, which worsens its prognosis. A diagnosis based on clinical symptoms alone is limited by the presence of other pulmonary changes that can manifest in a similar manner(2,22). On HRCT scans, increases in the caliber of the pulmonary artery trunk (normal value: 28.6 ± 2 mm) and main pulmonary arteries are often observed, although their absence does not exclude the diagnosis. A finding of pericardial effusion, especially in the anterior recess, with a thickness > 10 mm, is indicative of a poor prognosis and is also a strong predictor of pulmonary arterial hypertension on echocardiography(2326). Another pulmonary vascular manifestation in SSc is veno-occlusive disease, which is characterized by intimal proliferation and fibrosis of the intrapulmonary veins and venules, together with arteriolar involvement. The definitive diagnosis is obtained by biopsy, and alternatively can be performed clinically, in view of the increased risk of post-operative complications(22). Recent histological studies have shown that the venous involvement in the pulmonary form of SSc is greater than previously described, which explains, at least in part, the fact that patients with SSc-related pulmonary arterial hypertension are more likely to be refractory to treatment than are those with the idiopathic form(27). In patients with SSc-related pulmonary arterial hypertension, HRCT reveals centrilobular ground-glass attenuation, septal thickening, and lymph node enlargement(22). Other changes Some of the changes observed on CT scans of patients with SSc can be concomitant to findings of parenchymal disease and merit attention because of their frequency or severity. A common finding in patients with SSc, with a prevalence of 4174%, is lymphadenopathy, which appears to be associated with the presence of chronic interstitial lung disease secondary to an inflammatory response to aspiration pneumonia and reflux or other concomitant diseases such as lymphoma and sarcoidosis(24,28). Some studies have shown that the incidence of neoplastic diseases, especially those involving the lungs, is a higher among individuals with SSc. The overall incidence of neoplasms in SSc patients has been reported to be as high as 10.7%, and the risk of developing lung cancer is up to seven times greater among smokers with SSc than among other smokers(29). CONCLUSION Pulmonary complications arising from SSc can have a quite adverse clinical course, and the main clinical dilemma is related to the indication for and appropriate timing of immunosuppression therapy. Immunosuppressive agents play an important role in slowing the progression of fibrosis, and potential adverse effects related to such medication should be considered, being essential to the recognition of various radiological patterns of pulmonary disease. The changes most frequently observed in SSc are those that result from interstitial involvement, although manifestations related to alveolar and vascular involvement should always be borne in mind in cases in which the clinical picture is atypical and there is an inadequate response to the treatment, due to the need for an immediate, specific therapeutic approach. Other thoracic manifestations of SSc are also observed on CT scans of the chest and can be associated with lung disease. Such changes should also be recognized and correlated with the clinical findings in order to inform the handling of cases. REFERENCES 1. Varga J. Systemic sclerosis: an update. Bull NYU Hosp Jt Dis. 2008;66:198202. 2. Wells AU, Steen V, Valentini G. Pulmonary complications: one of the most challenging complications of systemic sclerosis. Rheumatology (Oxford). 2009;48 Suppl 3:iii404. 3. Chifflot H, Fautrel B, Sordet C, et al. Incidence and prevalence of systemic sclerosis: a systematic literature review. Semin Arthritis Rheum. 2008;37:22335. 4. Gohari Moghadam K, Gharibdoost F, Parastandechehr G, et al. Assessments of pulmonary involvement in patients with systemic sclerosis. Arch Iran Med. 2011;14:226. 5. Kim EA, Lee KS, Johkoh T, et al. Interstitial lung diseases associated with collagen vascular diseases: radiologic and histopathologic findings. Radiographics. 2002;22 Spec No:S15165. 6. Goh NS, du Bois RM. Interstitial disease in systemic sclerosis. In: Wells AU, Denton CP, editors. Handbook of systemic autoimmune diseases. 1st ed. Amsterdam, The Netherlands: Elsevier; 2004. p. 181207. 7. Wells AU. High-resolution computed tomography and scleroderma lung disease. Rheumatology (Oxford). 2008;47 Suppl 5:v5961. 8. King TE Jr. Nonspecific interstitial pneumonia and systemic sclerosis. Am J Respir Crit Care Med. 2002;165:15789. 9. D'Angelo WA, Fries JF, Masi AT, et al. Pathologic observations in systemic sclerosis (scleroderma): a study of fifty-eight autopsy cases and fifty-eight matched controls. Am J Med. 1969;46:42840. 10. Fujita J, Yoshinouchi T, Ohtsuki Y, et al. Non-specific interstitial pneumonia as pulmonary involvement of systemic sclerosis. Ann Rheum Dis. 2001;60:2813. 11. Bouros D, Wells AU, Nicholson AG, et al. Histopathologic subsets of fibrosing alveolitis in patients with systemic sclerosis and their relationship to outcome. Am J Respir Crit Care Med. 2002;165:15816. 12. Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188:73348. 13. American Thoracic Society/European Respiratory. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002;165:277304. 14. Taylor JG, Bolster MB. Bronchiolitis obliterans with organizing pneumonia associated with scleroderma and scleroderma spectrum diseases. J Clin Rheumatol. 2003;9:23945. 15. Shimizu Y, Tsukagoshi H, Nemoto T, et al. Recurrent bronchiolitis obliterans organizing pneumonia in a patient with limited cutaneous systemic sclerosis. Rheumatol Int. 2002;22:2168. 16. Bridges AJ, Hsu KC, Dias-Arias AA, et al. Bronchiolitis obliterans organizing pneumonia and scleroderma. J Rheumatol. 1992;19:113640. 17. Davison AG, Epstein O. Relapsing organising pneumonitis in a man with primary biliary cirrhosis, CREST syndrome, and chronic pancreatitis. Thorax. 1983;38:3167. 18. Azevedo ABC, Guimarães SMM, Tavares Júnior WC, et al. Avaliação da tomografia de alta resolução versus radiografia de tórax na doença intersticial pulmonar na esclerose sistêmica. Radiol Bras. 2005;38:959. 19. Ebert EC. Esophageal disease in scleroderma. J Clin Gastroenterol. 2006;40:76975. 20. Bhalla M, Silver RM, Shepard JA, et al. Chest CT in patients with scleroderma: prevalence of asymptomatic esophageal dilatation and mediastinal lymphadenopathy. AJR Am J Roentgenol. 1993;161:26972. 21. Le Pavec J, Girgis RE, Lechtzin N, et al. Systemic sclerosis-related pulmonary hypertension associated with interstitial lung disease: impact of pulmonary arterial hypertension therapies. Arthritis Rheum. 2011;63:245664. 22. Hassoun PM. Lung involvement in systemic sclerosis. Presse Med. 2011;40:e25e39. 23. Lynch DA. Lung disease related to collagen vascular disease. J Thorac Imaging. 2009;24:299309. 24. Baque-Juston MC, Wells AU, Hansell DM. Pericardial thickening or effusion in patients with pulmonary artery hypertension: a CT study. AJR Am J Roentgenol. 1999;172:3614. 25. Kuriyama K, Gamsu G, Stern RG, et al. CT-determined pulmonary artery diameters in predicting pulmonary hypertension. Invest Radiol. 1984;19:1622. 26. Fischer A, Misumi S, Curran-Everett D, et al. Pericardial abnormalities predict the presence of echocardiographically defined pulmonary arterial hypertension in systemic sclerosis-related interstitial lung disease. Chest. 2007;131:98892. 27. Dorfmüller P, Humbert M, Perros F, et al. Fibrous remodeling of the pulmonary venous system in pulmonary arterial hypertension associated with connective tissue diseases. Hum Pathol. 2007;38:893902. 28. Wechsler RJ, Steiner RM, Spirn PW, et al. The relationship of thoracic lymphadenopathy to pulmonary interstitial disease in diffuse and limited systemic sclerosis: CT findings. AJR Am J Roentgenol. 1996;167:1014. 29. Pontifex EK, Hill CL, Roberts-Thomson P. Risk factors for lung cancer in patients with scleroderma: a nested case-control study. Ann Rheum Dis. 2007;66:5513. 1. Adjunct Professor in the Department of Anatomy and Imaging at the Faculdade de Medicina da Universidade Federal de Minas Gerais (UFMG), Belo Horizonte, MG, Brazil 2. Associate Professor in the Department of Clinical Medicine at the Faculdade de Medicina da Universidade Federal de Minas Gerais (UFMG), Belo Horizonte, MG, Brazil 3. Adjunct Professor in the Department of Locomotor Studies at the Faculdade de Medicina da Universidade Federal de Minas Gerais (UFMG), Belo Horizonte, MG, Brazil Mailing address: Dra. Andréa de Lima Bastos Avenida Professor Alfredo Balena, 190, 1º andar, sala 179, Centro Belo Horizonte, MG, Brazil, 30130-100 E-mail: andrealb@ufmg.br Received June 14, 2015. Accepted after revision November 4, 2015. Study conducted at the Faculdade de Medicina da Universidade Federal de Minas Gerais (UFMG), Belo Horizonte, MG, Brazil. |
|
Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554