Radiologia Brasileira - Publicação Científica Oficial do Colégio Brasileiro de Radiologia
AMB - Associação Médica Brasileira CNA - Comissão Nacional de Acreditação
 Vol. 48 nº 3 - Maio / Jun. of 2015
Vol. 48 nº 3 - Maio / Jun. of 2015
|
ARTIGO DE REVISÃO
|
|
|
|
|
Autho(rs): Valdair F. Muglia1; Adilson Prando2 |
|
|
Descritores: Carcinoma de células renais; Câncer renal; Tomografia computadorizada; Ressonância magnética. |
|
|
Resumo: INTRODUÇÃO
Os carcinomas de células renais (CCRs) são o sétimo tipo histológico de câncer mais comum no mundo ocidental e vêm apresentando uma tendência mantida de aumento em sua prevalência(1,2). Os CCRs compreendem 1% a 3% de todas as neoplasias malignas viscerais. Aproximadamente 40% dos pacientes com CCR morrem devido à progressão da doença, tornando este tumor a lesão maligna urológica mais letal. Atualmente, a maioria dos CCRs é descoberta incidentalmente em exames de imagem realizados por razões urológicas ou não. Há um nítido predomínio do gênero masculino, que representa cerca de dois terços dos casos. A classificação histológica dos CCRs é de extrema importância, uma vez que a determinação dos subtipos histológicos tem significativas implicações prognósticas e terapêuticas(3,4). Como se sabe, o recente desenvolvimento da imuno-histoquímica fez com que a biópsia percutânea guiada por método de imagem se tornasse um procedimento minimamente invasivo, dotado de relativamente alta acurácia (70-90%) na caracterização histopatológica pré-operatória do tumor renal(5,6). Embora apenas minimamente invasiva, a biópsia percutânea tem riscos e limitações, sendo indicada, nos dias atuais, apenas nos casos em que a caracterização radiológica do tumor renal é impossível ou em situações clínicas específicas. Torna-se importante, então, a caracterização radiológica pré-operatória dos subtipos de CCRs, que, dependendo da situação clínica, pode ser complementada, ou não, pela biópsia percutânea confirmatória. Este procedimento é particularmente útil em pacientes sem adequadas condições cirúrgicas, com doença metastática, com rim único e lesão tumoral de difícil tratamento cirúrgico conservador e naqueles pacientes com indicação de receber terapia de alvo molecular(7-10). Há cerca de duas décadas, os CCRs eram divididos entre as variantes células claras e os do tipo células granulares(3). A Organização Mundial da Saúde (OMS), segundo a classificação de 2004, reconhece vários subtipos histológicos, que são descritos na Tabela 1. Os subtipos histológicos mais frequentes são células claras (CCRcc), papilífero (CCRp) e cromófobo (CCRcr) (Figura 1). Esses três, em conjunto, representam mais de 90% de todos os CCRs(11). 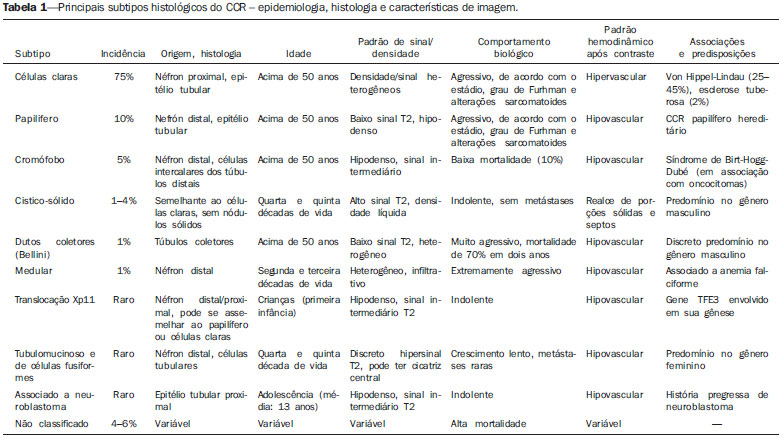 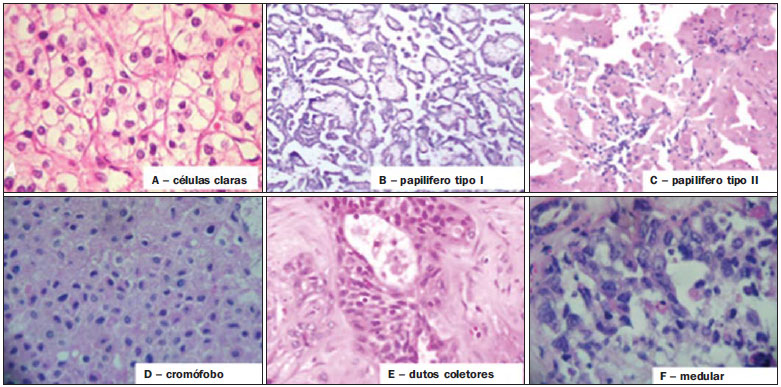 Figura 1. Histologia dos subtipos histológicos mais comuns. A: CCR de células claras - células com citoplasma amplo e rico em lipídios, conferindo nome à neoplasia. B: CCR papilífero tipo 1 - células basófilas, pequenas, com citoplasma escasso, organizadas em arranjo fusiforme, dispostas numa camada única ao redor da membrana basal. C: CCR papilífero tipo 2 - células dispostas em arranjo fusiforme, com papilas cobertas por células com abundante citoplasma eosinofílico, granular, com nucléolos proeminentes. D: CCR cromófobo - grandes células pálidas, com citoplasma reticulado e halos perinucleares. E: CCR de dutos coletores - histologia mostra arranjo de células irregulares, infiltrativas, na parede dos dutos coletores, com acentuada desmoplasia. F: CCR medular - neoplasia originária em nefrón distal também com arranjo de células irregulares, acentuado pleomorfismo e núcleos hipercromáticos. Em 2013, a Sociedade Internacional de Uropatologia (ISUP) propôs uma nova classificação do CCR, incluindo a proposta da OMS, porém sugerindo cinco novos tipos de neoplasia renal bem caracterizados e três mais que foram considerados como entidades novas e emergentes(12). Os exames de imagem têm papel importante no diagnóstico dos CCRs, sendo responsáveis pela tendência em se estabelecer o diagnóstico em estádios mais precoces, além de serem essenciais no estadiamento e planejamento terapêutico(13). Recentemente, trabalhos têm mostrado que os métodos de imagem podem separar os carcinomas da variante células claras dos tipos histológicos papilíferos e cromófobos, respectivamente, segundo e terceiro tipos mais comuns(14). Além das técnicas tradicionais, com avaliação da densidade, intensidade de sinal e padrão de impregnação das lesões pelo meio de contraste intravenoso, outras técnicas, principalmente as quantitativas por ressonância magnética (RM), como difusão e perfusão, têm sido empregadas na caracterização das neoplasias epiteliais renais. O objetivo deste artigo é revisar as principais características histológicas e os aspectos de imagem encontrados nas principais variantes histológicas do CCR, correlacionando-os com casos clínicos de CCRs das instituições dos autores. CARCINOMA DE CÉLULAS CLARAS Esta variante é a mais comum, representando entre 70% e 75% de todos os CCRs(3,7). Na grande maioria é do tipo esporádico (95%), e nos 5% restantes, associados a síndromes hereditárias (von Hippel-Lindau, esclerose tuberosa). O CCRcc se origina do epitélio dos túbulos contornados proximais (córtex renal) e apresenta crescimento predominantemente expansivo. Macroscopicamente, é uma lesão sólida, amarelada, com graus variáveis de necrose, hemorragia e degeneração cística internas, sendo esses achados mais comuns nos tumores de grande volume e com crescimento rápido. Calcificações tumorais também podem ser encontradas. Histologicamente, estas lesões apresentam células claras devido ao seu conteúdo citoplasmático rico em lipídios e glicogênio(4). Estes tumores frequentemente apresentam também células com citoplasma granular eosinofílico. Os achados de imagem refletem estes aspectos histopatológicos, identificando-se lesões hipervascularizadas e heterogêneas, devido a necrose, hemorragia, cistos e calcificações. A necrose é mais comum nas lesões maiores, em geral de dimensões acima de 4 cm. A porcentagem de ocorrência e o grau de necrose também têm sido associados a tumores com histologia de alto grau(15,16). Na tomografia computadorizada (TC), estes tumores apresentam, usualmente, impregnação intensa por contraste na fase corticomedular (120-140 UH) e característica desimpregnação (lavagem) na fase nefrográfica (90-100 UH) (Figura 2). 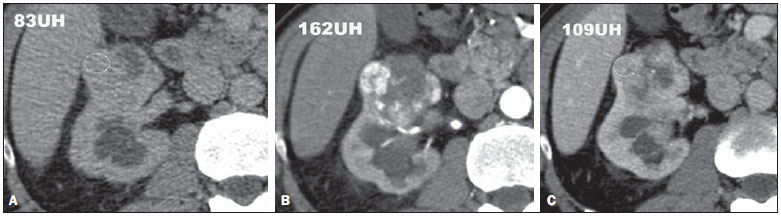 Figura 2. CCR variante células claras. TC nas fases pré-contraste (A), corticomedular (B) e nefrográfica (C). Notar lesão expansiva heterogênea com componentes císticos internos e áreas sólidas periféricas com densidade média de 83 UH na fase pré-contraste, fortemente impregnável por contraste, passando para 162 UH na fase corticomedular e para 109 UH na fase nefrográfica. Na RM, estas lesões têm intensidade de sinal semelhante à do córtex renal nas imagens ponderadas em T1 e hipersinal nas imagens ponderadas em T2. Em razão do conteúdo de lipídio intracelular, outro achado encontrado em até 60% dos CCRcc é a presença da perda de sinal nas imagens em oposição de fase (out of phase) da técnica chemical shift imaging(17) (Figura 3). Devemos lembrar que este achado não é específico do CCRcc, pois pode ser observado também em angiomiolipoma sem gordura macroscópica e, mais raramente, também no CCRp. Os CCRcc podem apresentar também uma pseudocápsula hipodensa na fase corticomedular da TC e com hipossinal em T1 e T2 na RM. Descontinuidade desta pseudocápsula usualmente indica um tumor de alto grau. A análise comparativa da intensidade de sinal das lesões na fase corticomedular da RM com contraste intravenoso é uma ferramenta efetiva na diferenciação entre CCRcc e CCRp. Em uma análise quantitativa da impregnação tumoral no exame multifásico (fases corticomedular, nefrográfica e excretora), evidenciou-se que a porcentagem de alteração de sinal nestas três fases após meio de contraste em relação à fase pré-contraste foi significativamente maior no CCRcc (230%, 250% e 227% para as fases corticomedular, nefrográfica e excretora, respectivamente) do que no CCRp (49%, 92% e 88% para as fases corticomedular, nefrográfica e excretora, respectivamente) ou no CCRcr (98%, 183% e 159% para as fases corticomedular, nefrográfica e excretora, respectivamente)(14). 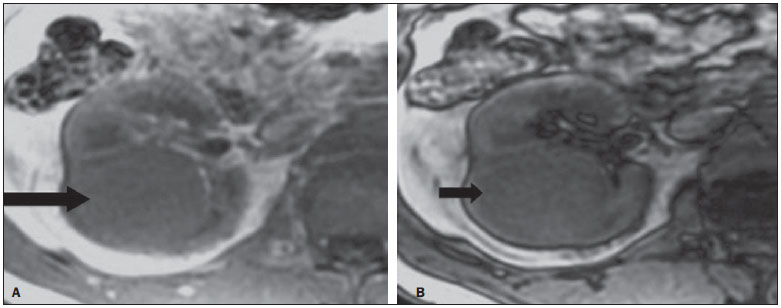 Figura 3. CCR variante células claras. RM, técnica de chemical shift imaging, imagens em fase (A) e fora de fase (B) mostrando lesão expansiva sólida no rim direito (setas), que apresenta ligeira queda de sinal na sequência fora de fase, de difícil percepção visual, caracterizada apenas pela medida do índice de queda de sinal da lesão de 11% (índice de queda de sinal = intensidade de sinal em fase intensidade de sinal fora de fase / intensidade de sinal em fase × 100). Embora ainda com resultados controversos, de maneira geral o emprego de imagens ponderadas em difusão, pela RM, não tem sido até o momento útil na diferenciação entre os subtipos histológicos dos CCRs(18). Outro achado relativamente comum destas lesões é a extensão para a gordura perirrenal, para a gordura do seio renal e para o interior das veias renal e cava inferior. Na vigência de invasão venosa, é comum a presença de circulação venosa colateral perirrenal. Metástases hematogênicas são relativamente comuns nos carcinomas de células claras e comprometem principalmente pulmão, fígado e ossos(19). Metástases linfonodais são descritas em até 15% dos casos. Como regra geral, o CCRcc tem um prognóstico pior do que os outros dois subtipos histológicos mais comuns, o CCRp e o CCRcr(4,11). CARCINOMA PAPILÍFERO A variante papilífera é o segundo subtipo histológico mais comum dos CCRs, representando cerca de 10% nas diversas séries descritas na literatura(3,4). Também pode ocorrer esporadicamente ou em condições familiares. Esta variante apresenta dois subtipos baseados na sua aparência histológica e comportamento biológico. Esses dois subtipos têm prognósticos bastante distintos, porém, pelas atuais técnicas de imagem, não é possível distingui-los na fase pré-operatória(20). Os achados histológicos incluem células organizadas em arranjo fusiforme, podendo apresentar áreas de hemorragia interna e alterações císticas, principalmente nas lesões maiores(11). Os CCRp do tipo 1 (basófilo) apresentam células basófilas dispostas em camada única ao redor da membrana basal. Essas pequenas células têm escasso citoplasma claro e núcleos hipercromáticos. Já o subtipo histológico 2 (eosinofílico) apresenta papilas cobertas por células com abundante citoplasma eosinofílico, granular, com nucléolos proeminentes associados a áreas de necrose. Os CCRp têm, habitualmente, melhor prognóstico que os CCRcc. O CCRp tipo 1, tipicamente, apresenta-se em estádio e grau mais baixos que o CCRp tipo 2, sendo, portanto, associado a um melhor prognóstico. O CCRp do tipo 2, habitualmente, à época do diagnóstico, apresenta-se como neoplasia de alto grau, frequentemente associada a metástases ganglionares e, às vezes, a invasão venosa. Os CCRp tendem a ser lesões sólidas, bem definidas e com crescimento lento(21). Possuem tendência a bilateralidade (4%) e/ou multifocalidade (22,5%) e estão associados a doença renal crônica adquirida. Na TC e na RM, esses tumores tendem a ser mais homogêneos que o CCRcc, e hipovascularizados, quando comparados ao parênquima renal adjacente(15,16,22). Na fase corticomedular do estudo por TC, com contraste intravenoso, tendem a apresentar densidade média de 50-60 UH, passando para uma densidade média de 65-75 UH na fase nefrográfica (impregnação progressiva). Nas imagens por RM, os carcinomas papilíferos se manifestam comumente como lesões periféricas, com intenso hipossinal em T2 (Figura 4), presumivelmente devido ao conteúdo de hemossiderina intratumoral(23) ou ao seu arranjo arquitetural. Lesões maiores que 4,0 cm de diâmetro podem ser heterogêneas devido à presença de necrose, hemorragia e calcificação. Apenas raramente podem apresentar focos internos de gordura macroscópica (macrófagos intersticiais com colesterol) ou mesmo gordura intracitoplasmática, e neste caso, apresentar-se também com queda de sinal na sequência fora de fase, na técnica de chemical shift imaging. 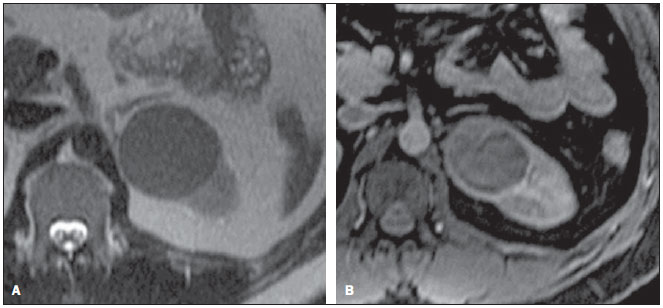 Figura 4. CCR variante papilífero. A: RM, corte axial em T2 mostrando lesão expansiva homogênea, com intenso hipossinal. B: RM, corte axial T1 após meio de contraste intravenoso mostrando lesão acentuadamente hipovascular em relação ao córtex adjacente. Os CCRp também podem se apresentar como lesões neoplásicas císticas, usualmente com conteúdo hemorrágico e projeções sólidas papilíferas murais hipovascularizadas. CARCINOMA DE CÉLULAS RENAIS DO TIPO CROMÓFOBO O CCRcr representa cerca de 5% de todas as neoplasias epiteliais renais malignas, sendo mais frequente na sexta década de vida. Este subtipo é menos agressivo que o CCRcc(3,11), apresentando o melhor prognóstico entre os CCRs. Metástases ocorrem em apenas 7% dos pacientes. Patologicamente, esta variante menos agressiva tem coloração alaranjada, mudando para cinza, após fixação. À microscopia, apresentam grandes células pálidas, com citoplasma reticulado e halos perinucleares. Quando ocorre transformação sarcomatoide, as lesões são mais agressivas e o prognóstico é consideravelmente pior(3,24). Alguns patologistas acreditam que exista uma relação muito estreita entre a variante cromófoba e os oncocitomas. Ambos seriam derivados de células intercaladas nos dutos coletores e são achados comuns, associados, na síndrome de Birt-Hogg-Dubé(25). Nos exames de imagem, os carcinomas cromófobos tendem a ser mais homogêneos que os de células claras(13,14,21). Essas lesões são hipovascularizadas em relação ao córtex renal, apresentando, na maioria das vezes, moderada impregnação por contraste (80-100 UH na fase corticomedular do exame tomográfico), sendo, portanto, menos intensa que a variante de células claras e mais intensa quando comparados com a variante papilífera(21) (Figura 5). Não raro, todavia, o CCRcr pode apresentar-se como lesão homogênea e fortemente vascularizada na fase corticomedular (120-140 UH). Na RM, a lesão tende a apresentar leve hipossinal ou sinal intermediário nas imagens ponderadas em T2 (Figura 6), podendo inclusive apresentar cicatriz central. Deve-se ressaltar que os achados do CCRcr são indistinguíveis dos oncocitomas (tumores benignos originados das células intercaladas, do tipo B, dos dutos coletores corticais), os quais podem, inclusive, apresentar também cicatriz central. Necrose pode ocorrer nas lesões volumosas. 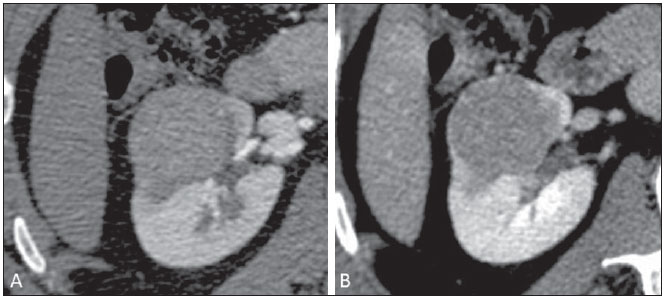 Figura 5. CCR variante cromófobo. TC nas fases corticomedular (A) e nefrográfica (B) mostrando lesão expansiva sólida homogeneamente impregnável por contraste, com densidade média de 92 UH e 126 UH, respectivamente. 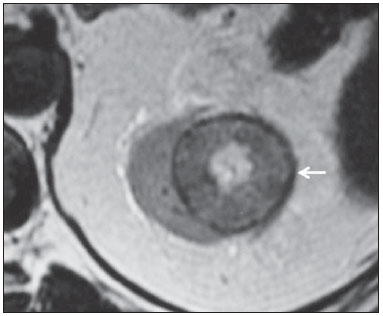 Figura 6. CCR variante cromófobo. RM, plano axial, imagem ponderada em T2 identificando lesão expansiva bem delimitada no rim esquerdo, com sinal intermediário nesta ponderação, nítida pseudocápsula (seta) e área de cicatriz central. CARCINOMA DOS DUTOS COLETORES (TUMOR DE BELLINI) Esta rara variante de CCR representa menos de 1% de todas as neoplasias malignas do rim, porém, sua importância reside no fato de ser um tumor altamente agressivo(3,9,26). Apresenta prognóstico bastante desfavorável, pois 30% dos pacientes já apresentam metástases ao diagnóstico e 60-70% deles morrem dentro de um período de dois anos(27). Histologicamente, o carcinoma de células de dutos coletores (CCDC) consiste de um arranjo de células irregulares, infiltrativas, na parede dos dutos coletores, exibindo acentuada desmoplasia(13). Há discreto predomínio do sexo masculino e, em geral, essas lesões aparecem depois da quinta década de vida. Nos exames de imagem, os CCDCs apresentam-se como lesões heterogêneas com sinal extremamente variável em T2, dependendo da quantidade de hemorragia, necrose, componente cístico e calcificação, e com frequência apresentam hipossinal nesta ponderação (Figura 7). Em geral, as lesões iniciam-se na medula, sendo uma característica desses tumores e que implica o diagnóstico diferencial com os carcinomas de células transicionais(28). As lesões maiores podem invadir o córtex. A maioria dessas lesões é sólida, porém algumas podem se apresentar como lesões complexas(26). São hipovascularizadas tanto na TC de múltiplos detectores como na RM e possuem a tendência de impregnação por contraste heterogênea ou periférica. 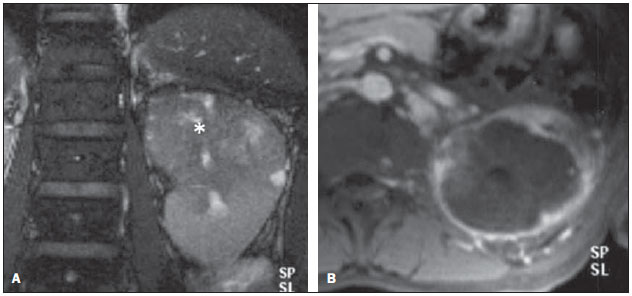 Figura 7. CCR variante dutos coletores. A: RM, plano coronal, ponderação T2 mostrando lesão expansiva irregular no polo superior do rim esquerdo, com sinal heterogêneo e predomínio de hipossinal (asterisco). B: RM, ponderação T1 após meio de contraste, no plano axial a lesão apresenta realce heterogêneo, predominantemente periférico, bem menos intenso que o córtex renal. CARCINOMA DE CÉLULAS RENAIS CÍSTICO MULTILOCULAR É uma lesão muito infrequente, com incidência que varia entre 1-4% de todos os CCRs(29) e que ocorre mais frequentemente em homens, em uma proporção de 3:1, com média de idade de cerca de 50 anos. Apresenta-se como lesão cística multisseptada, separada do rim por uma cápsula fibrosa(16). Tem excelente prognóstico e pode ser totalmente curado. Recorrência e metástases não têm sido descritas. Ao exame histopatológico encontram-se cistos serosos, gelatinosos ou hemorrágicos, revestidos por única camada de pequenas coleções de células epiteliais com citoplasma claro. Não existe, neste tumor, a formação de nódulos sólidos, achado que distingue o CCR cístico multilocular do CCRcc com extensa degeneração cística(30). Calcificação nos septos ou na pseudocápsula ocorre em até 20% dos casos. Na TC e na RM manifestam-se como cistos complexos, assumindo, na classificação de Bosniak, desde categoria II-F a categoria IV(16) (Figura 8). 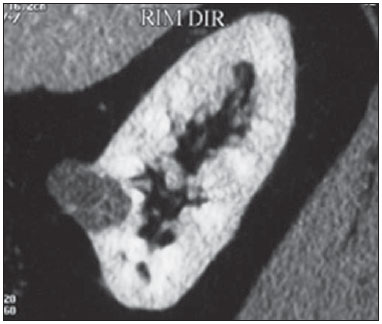 Figura 8. CCR cístico multilocular. Reconstrução sagital da fase nefrográfica da TC mostrando a natureza cística multilocular da lesão. CARCINOMA DE CÉLULAS RENAIS VARIANTE MEDULAR Este raro tipo de variante do CCR foi descrito inicialmente em 1995 por Davis. Alguns patologistas consideram esta variante um subtipo do CCDC. Além de muito rara(31), é uma neoplasia extremamente agressiva. Origina-se no néfron distal, nas células de dutos coletores. Uma característica marcante deste tipo histológico é sua associação com portadores de anemia falciforme e traço falcêmico. Em decorrência desta associação, caracteristicamente acomete pacientes jovens na segunda e terceira décadas de vida. Aos exames de imagem apresentam-se como lesões infiltrativas que promovem obstrução e dilatação de cálices. São lesões hipovascularizadas (Figura 9) e frequentemente associadas a adenopatia locorregional(32). 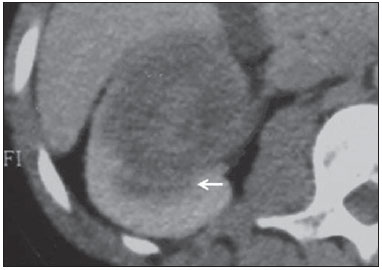 Figura 9. CCR variante medular. Paciente do sexo masculino, 25 anos de idade, com anemia falciforme. TC com contraste intravenoso mostrando extensa lesão expansiva sólida, hipovascularizada, predominantemente medular e discretamente heterogênea, no rim direito. Notar o aspecto infiltrativo da lesão no sistema pielocalicinal (seta) e na porção proximal do ureter. CARCINOMA DE CÉLULAS RENAIS VARIANTE TUBULOMUCINOSO E DE CÉLULAS FUSIFORMES (CTMCF) Esta neoplasia renal muito rara, recentemente descrita, origina-se de células de túbulo contorcido distal e dutos coletores. Apresenta nítido predomínio para o gênero feminino, usualmente na quarta e quinta décadas de vida(3,11). É considerada uma neoplasia de baixo grau e, dos poucos casos descritos até hoje na literatura, nenhum apresentou proliferação. À histologia, é composto por células densamente agrupadas, intercaladas por estroma mucinoso claro e um componente de células fusiformes bem definido(4). Nos exames de imagem o CTMCF não tem uma apresentação característica, e os poucos casos relatados até o momento apresentam-se como nódulos ou massas circunscritas, incidentalmente descobertos em exames de ultrassonografia e TC. Tipicamente, tendem a ser homogêneos com sinal baixo em T1 e intermediário a discretamente hiperintenso em T2(22). Algumas dessas lesões podem apresentar uma cicatriz central irregular. A neoplasia apresenta-se hipovascularizada em relação ao córtex adjacente, após a injeção de meio de contraste intravenoso, e quando presente, a cicatriz central (Figura 10) pode exibir graus variáveis de opacificação(33). 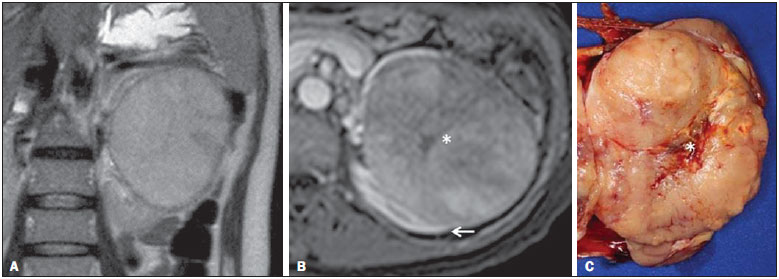 Figura 10. CCR variante tubulomucionoso e de células fusiformes. Paciente do sexo feminino, 57 anos de idade, com hematúria. A: RM na ponderação coronal T2 mostrando lesão expansiva com sinal intermediário e (B) hipovascular após meio de contraste, em fase nefrográfica comparar com o córtex (seta). Notar a cicatriz de hipossinal (asterisco). Apesar das grandes dimensões, a lesão é bem delimitada e sem aspecto infiltrativo. C: Espécime cirúrgico mostrando lesão circunscrita, de coloração amarelada, com cicatriz central (asterisco). CARCINOMA DE CÉLULAS RENAIS ASSOCIADO A TRANSLOCAÇÃO DO BRAÇO CURTO DO CROMOSSOMO Este é um tipo bastante raro de neoplasia, recentemente descrito, com sua origem envolvendo o gene TFE3 e translocações envolvendo o braço curto do cromossomo do 11 Xp11.2(3,4). Essa neoplasia é vista, em geral, em crianças e adultos jovens. Nos achados patológicos, à macroscopia lembra um CCRcc e se apresenta como massa bem delimitada com pseudocápsula; no entanto, histologicamente, apresenta projeções papilíferas que lembram a neoplasia de mesmo nome. Tem prognóstico mais indolente em comparação à variante células claras(34). Não há achados de imagem característicos para essas lesões, que em geral podem se apresentar como lesões heterogêneas, principalmente as maiores, e hipervascularizadas(35). CARCINOMA DE CÉLULAS RENAIS ASSOCIADO A NEUROBLASTOMA Esta também é uma neoplasia excepcionalmente rara. Há apenas alguns casos descritos na literatura, em crianças que a desenvolveram após terem sido tratadas de neuroblastoma(3,36). Pode ser uni ou bilateral e não há predileção de gênero. Como surge após o tratamento do neuroblastoma, em geral acomete pacientes jovens com idade média de 13 anos. Esse tipo de neoplasia, nos raros casos descritos, apresenta-se como lesão bem delimitada, hipovascularizada, em geral de comportamento mais indolente(34). CARCINOMAS RENAIS NÃO CLASSIFICADOS Mesmo com a classificação de 2004, em 4-6% das neoplasias renais não é possível, ainda, uma caracterização histológica precisa(3,5), o que poderá ser melhorado com a classificação de Vancouver proposta pela ISUP(12). Os tumores desta categoria são histologicamente heterogêneos e, na maioria das vezes, de alto grau. Alguns achados que servem para identificá-los são: presença de componente sarcomatoide sem elementos epiteliais reconhecidos; produção de mucina; mistura de elementos estromais e epiteliais; e tipos celulares não reconhecidos(37). Nesta categoria encontram-se os subtipos histológicos de CCR com pior prognóstico. CONCLUSÃO A classificação histológica dos CCRs é de extrema importância, uma vez que a determinação dos subtipos histológicos tem significativa implicação prognóstica e terapêutica. Torna-se importante o esforço do radiologista na tentativa de reconhecimento pré-operatório (por imagem) dos subtipos histológicos do CCR. Atualmente, na prática clínica diária, os melhores resultados estão sendo obtidos na diferenciação entre CCRs de células claras e CCRs não células claras. Aguardam-se resultados de novas pesquisas e estudos prospectivos que adicionem novas informações ao estado atual. REFERÊNCIAS 1. Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71-96. 2. Ljunberg B, Campbell SC, Cho HY, et al. The epidemiology of renal cell carcinoma. Eur Urol. 2011;60:615-21. 3. Lopez-Beltran A, Scarpelli M, Montironi R, et al. 2004 WHO classification of renal tumours of the adults. Eur Urol. 2006;49:798-805. 4. Decastro GJ, McKiernan JM. Epidemiology, clinical staging, and presentation of renal cell carcinoma. Urol Clin North Am. 2008;35:581-92. 5. Dechet CB, Zincke H, Sebo TJ, et al. Prospective analysis of computerized tomography and needle biopsy with permanent sectioning to determine the nature of solid renal masses in adults. J Urol. 2003;169:71-4. 6. Silverman SG, Gan YU, Mortele KJ, et al. Renal masses in the adult patient: the role of percutaneous biopsy. Radiology. 2006;240:6-22 7. Upton MP, Parker RA, Youmans A, et al. Histologic predictors of renal cell carcinoma response to interleukin-2-based therapy. J Immunother. 2005;28:488-95. 8. Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115-24. 9. Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125-34. 10. Dutcher JP, Szczylik C, Tannir N, et al. Correlation of survival with tumor histology, age, and prognostic risk group for previously untreated patients with advanced renal cell carcinoma (adv RCC) receiving temsirolimus (TEMSR) or interferon-alpha (IFN) [Abstract]. J Clin Oncol. 2007;25(Suppl 18):5033. 11. Lopez-Beltran A, Carrasco JC, Cheng L, et al. 2009 update on the classification of renal epithelial tumors in adults. Int J Urol. 2009;16:432-43. 12. Delahunt B, Srigley JR, Montironi R, et al. Advances in renal neoplasia: recommendations from the 2012 International Society of Urological Pathology Consensus Conference. Urology. 2014;83:969-74. 13. Catalano C, Fraioli F, Laghi A, et al. High-resolution multidetector CT in the preoperative evaluation of patients with renal cell carcinoma. AJR Am J Roentgenol. 2003;180:1271-7. 14. Vargas HA, Chaim J, Lefkowitz RA, et al. Renal cortical tumors: use of multiphasic contrast-enhanced MR imaging to differentiate benign and malignant histologic subtypes. Radiology. 2012;264:779-88. 15. Pedrosa I, Sun MR, Spencer M, et al. MR imaging of renal masses: correlation with findings at surgery and pathologic analysis. Radiographics. 2008;28:985-1003. 16. Prasad SR, Humphrey PA, Catena JR, et al. Common and uncommon histologic subtypes of renal cell carcinoma: imaging spectrum with pathologic correlation. Radiographics. 2006;26:1795-806. 17. Prando A. Intratumotal fat in a renal cell carcinoma. AJR Am J Roentgenol. 1991;156:871. 18. Sandrasegaran K, Sundaram CP, Ramaswamy R, et al. Usefulness of diffusion-weighted imaging in the evaluation of renal masses. AJR Am J Roentgenol. 2010;194:438-45. 19. Feltrin LT, Ferreira JR, Mamere AE, et al. Signs of hypervascularization at magnetic resonance imaging in bone metastases from renal cell carcinoma. Radiol Bras. 2009;42:155-7. 20. Yoshimitsu K, Kakihara D, Irie H, et al. Papillary renal carcinoma: diagnostic approach by chemical shift gradient-echo and echo-planar MR imaging. J Magn Reson Imaging. 2006;23:339-44. 21. Lubensky IA, Schmidt L, Zhuang Z, et al. Hereditary and sporadic papillary renal carcinomas with c-met mutations share a distinct morphological phenotype. Am J Pathol. 1999;155:517-26. 22. Egbert ND, Caoili EM, Cohan RH, et al. Differentiation of papillary renal cell carcinoma subtypes on CT and MRI. AJR Am J Roentgenol. 2013;201:347-55. 23. Karlo CA, Donati OF, Burger IA, et al. MR imaging of renal cortical tumours: qualitative and quantitative chemical shift imaging parameters. Eur Radiol. 2013;23:1738-44. 24. Liu L, Qian J, Singh H, et al. Immunohistochemical analysis of chromophobe renal cell carcinoma, renal oncocytoma, and clear cell carcinoma: an optimal and practical panel for differential diagnosis. Arch Pathol Lab Med. 2007;131:1290-7. 25. Prando A, Prando D, Prando P. Renal cell carcinoma: unusual presentations. Radiographics. 2006;26:233-44. 26. Srigley JR, Eble JN. Collecting duct carcinoma of kidney. Semin Diagn Pathol. 1998;15:54-67. 27. Outwater EK, Bhatia M, Siegelman ES, et al. Lipid in renal clear cell carcinoma: detection on opposed-phase gradient-echo MR images. Radiology. 1997;205:103-7. 28. Orsola A, Trias I, Raventós CX, et al. Renal collecting (Bellini) duct carcinoma displays similar characteristics to upper tract urothelial cell carcinoma. Urology. 2005;65:49-54. 29. Murad T, Komaiko W, Oyasu R, et al. Multilocular cystic renal cell carcinoma. Am J Clin Pathol. 1991;95:633-7. 30. Eble JN, Bonsib SM. Extensively cystic renal neoplasms: cystic nephroma, cystic partially differentiated nephroblastoma, multilocular cystic renal cell carcinoma, and cystic hamartoma of renal pelvis. Semin Diagn Pathol. 1998;15:2-20. 31. Liu Q, Galli S, Srinivasan R, et al. Renal medullary carcinoma: molecular, immunohistochemistry, and morphologic correlation. Am J Surg Pathol. 2013;37:368-74. 32. Khan A, Thomas N, Costello B, et al. Renal medullary carcinoma: sonographic, computed tomography, magnetic resonance and angiographic findings. Eur J Radiol. 2000;35:1-7. 33. Lima MS, Barros-Silva GE, Pereira RA, et al. The imaging and pathological features of a mucinous tubular and spindle cell carcinoma of the kidney: a case report. World J Surg Oncol. 2013;11:34. 34. Srigley JR, Delahunt B. Uncommon and recently described renal carcinomas. Mod Pathol. 2009;22 Suppl 2:S2-S23. 35. Koo HJ, Choi HJ, Kim MH, et al. Radiologic-pathologic correlation of renal cell carcinoma associated with Xp11.2 translocation. Acta Radiol. 2013;54:827-34. 36. Fleitz JM, Wootton-Gorges SL, Wyatt-Ashmead J, et al. Renal cell carcinoma in long-term survivors of advanced stage neuroblastoma in early childhood. Pediatr Radiol. 2003;33:540-5. 37. Cheville JC, Lohse CM, Zincke H, et al. Comparisons of outcome and prognostic features among histologic subtypes of renal cell carcinoma. Am J Surg Pathol. 2003;27:612-24. 1. Pós-doutorado, Professor Associado do Centro de Ciências das Imagens e Física Médica (CCIFM) da Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo (FMRP-USP), Ribeirão Preto, SP, Brasil 2. Professor Convidado da Universidade Estadual de Campinas (Unicamp), Chefe do Departamento de Imaginologia do Hospital Vera Cruz, Campinas, SP, Brasil Endereço para correspondência: Dr. Valdair Francisco Muglia Hospital das Clínicas, FMRP-USP - Centro de Ciências das Imagens e Física Médica, 2º andar Avenida Bandeirantes, 3900, Monte Alegre Ribeirão Preto, SP, Brasil, 14048-900 E-mail: fmuglia@fmrp.usp.br Recebido para publicação em 21/9/2013. Aceito, após revisão, em 10/4/2014. Trabalho realizado na Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo (FMRP-USP), Ribeirão Preto, SP, Brasil. |
|
Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554