Radiologia Brasileira - Publicação Científica Oficial do Colégio Brasileiro de Radiologia
AMB - Associação Médica Brasileira CNA - Comissão Nacional de Acreditação
 Vol. 47 nº 2 - Mar. / Abr. of 2014
Vol. 47 nº 2 - Mar. / Abr. of 2014
|
QUAL É O SEU DIAGNÓSTICO?
|
|
|
|
|
Autho(rs): Alexandre Dias Mançano1; Rubens Carneiro dos Santos Neto2; Karla Coelho Caixeta e Silva3 |
|
|
Homem, 62 anos de idade, deu entrada no pronto-socorro com queixa de tosse e desconforto respiratório. Negava febre. Referia ter tosse crônica, que há cinco anos tem apresentado piora, com crises de desconforto respiratório que antes não tinha. Segundo o paciente, desde criança tem asma e crises de "bronquite" que pioram aos esforços físicos, mudança climática e contato com poeira e mofo. O paciente é ex-tabagista, tendo fumado durante 26 anos, e há 20 anos não fuma mais. Negava acompanhamento médico regular ou tratamentos prévios. Sempre procura somente o pronto-socorro, onde já ficou internado para tratamento de algumas pneumonias. Ao exame físico, apresentava estertores crepitantes nas bases pulmonares e sibilos.
Foi solicitada radiografia de tórax, que demonstrou opacidades reticulares esparsas, mais extensas no lobo inferior direito, além de bronquiectasias. Para melhor investigação do caso, foi solicitada tomografia computadorizada do tórax (Figuras 1 e 2). 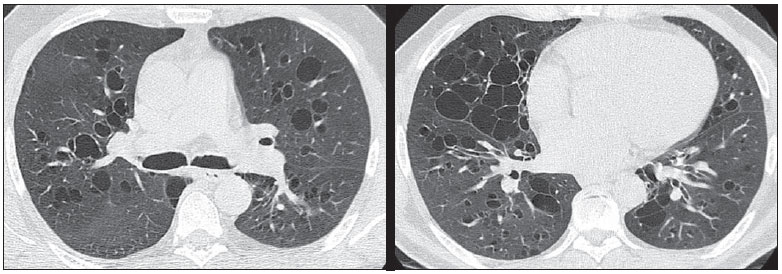 Figura 1. Tomografia computadorizada de alta resolução, com cortes nas regiões pulmonares médias e inferiores. 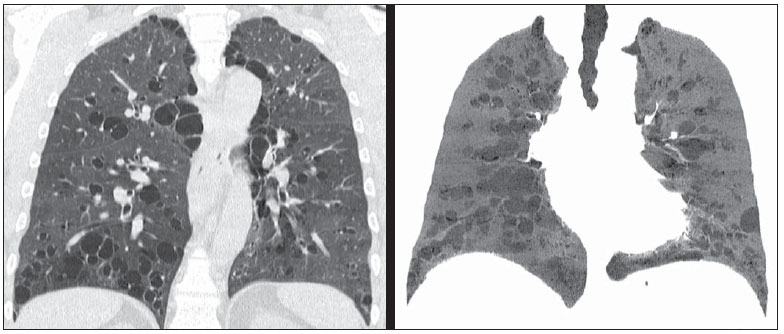 Figura 2. Tomografia computadorizada com reconstruções coronais e MINIP. Descrição das imagens Figuras 1 e 2. Tomografias computadorizadas do tórax mostram bronquiectasias císticas com paredes finas difusamente distribuídas pelos pulmões. Traqueia, brônquios principais e lobares preservados. Diagnóstico: Síndrome de Williams-Campbell. COMENTÁRIOS A síndrome de Williams-Campbell é uma doença rara, congênita, causada por alteração da cartilagem em brônquios subsegmentares, levando a bronquiectasias e colapso das vias aéreas distais(1,2). A deficiência dos anéis de cartilagem nos brônquios de quarta a sexta ordem resulta em bronquiectasias distais, com preservação da traqueia e brônquios principais(3). A síndrome é comumente descrita em crianças com história de pneumonias recorrentes e sintomas obstrutivos, sendo ocasionalmente diagnosticada na fase adulta(2-4). Há descrição de ocorrência familiar, sugerindo a necessidade de investigação dos familiares(1,5). O quadro clínico é de tosse, sibilos e pneumonias recorrentes(1,5). As provas de função respiratória demonstram um padrão obstrutivo, sendo que em casos avançados podem apresentar padrão misto. A deficiência da cartilagem altera a fisiologia brônquica, promovendo dilatação durante a inspiração e colabamento durante a expiração, determinando hiperinsuflação ou colabamento segmentar/lobar(3,5). Essas alterações levam ao aprisionamento aéreo, prejudicando a drenagem aérea e acúmulo de secreção(3,4). A recorrente destruição da árvore brônquica e drenagem mucoide inadequada resultam em destruição parenquimal distal à bronquiectasia. A gravidade dos sintomas e o prognóstico da doença dependem do grau e extensão do comprometimento da cartilagem(2). O aspecto de imagem na radiografia convencional é de bronquiectasias simétricas. Na tomografia computadorizada de alta resolução são observadas bronquiectasias císticas, distais, em brônquios de quarta a sexta ordem, de extensão variável, sem acometer traqueia ou brônquios principais(2). A preservação proximal da árvore brônquica é característica da síndrome e é o principal achado de diferenciação com a síndrome de Mounier-Kuhn (traqueobronquiomalácia)(5,6). Notam-se, ainda, impactação mucoide em alguns brônquios, predominantemente nos lobos inferiores, e achados concomitantes de doença infecciosa, caracterizados por padrão de árvore em brotamento. Pode ainda ser observado o padrão de perfusão em mosaico(1). O diagnóstico é feito pelos achados clínicos e tomográficos, complementados por história pregressa e familiar, além de exames laboratoriais para descartar outras hipóteses. Não há achados significativos na broncoscopia(1,2). O diagnóstico diferencial principal é a síndrome de Mounier-Kuhn (traqueobronquiomalácia), além de deficiência de alfa-1 antitripsina, fibrose cística, aspergilose broncopulmonar alérgica e síndrome de discinesia ciliar(1,2,4). A síndrome de Williams-Campbell não dispõe de tratamento específico, sendo a profilaxia e tratamento das exacerbações a principal forma de tratamento, baseando-se na fisioterapia respiratória e antibioticoterapia. Há descrição de alguns casos na literatura de transplante pulmonar, sem grande sucesso, sendo que um deles teve como achado pósmorte broncomalácia pós-transplante(1,3,7). REFERÊNCIAS 1. Konoglou M, Porpodis K, Zarogoulidis P, et al. Williams-Campbell syndrome: a case report. Int J Gen Med. 2012;5:41-4. 2. Di Scioscio V, Zompatori M, Mistura I, et al. The role of spiral multidetector dynamic CT in the study of Williams-Campbell syndrome. Acta Radiol. 2006;47:798-800. 3. George J, Jain R, Tariq SM. CT bronchoscopy in the diagnosis of Williams-Campbell syndrome. Respirology. 2006;11:117-9. 4. Cantin L, Bankier AA, Eisenberg RL. Bronchiectasis. AJR Am J Roentgenol. 2009;193:W158-71. 5. McAdams HP, Erasmus J. Chest case of the day. Williams-Campbell syndrome. AJR Am J Roentgenol. 1995;165:190-1. 6. Marom EM, Goodman PC, McAdams HP. Diffuse abnormalities of the trachea and main bronchi. AJR Am J Roentgenol. 2001;176:713-7. 7. Burguete SR, Levine SM, Restrepo MI, et al. Lung transplantation for Williams-Campbell syndrome with a probable familial association. Respir Care. 2012;57:1505-8. 1. Médico Radiologista da Radiologia Anchieta Hospital Anchieta, Coordenador da Residência Médica do Hospital Regional de Taguatinga, Taguatinga, DF, Brasil 2. Médico Estagiário da Radiologia Anchieta Hospital Anchieta, Taguatinga, DF, Brasil 3. Médica Residente do Hospital Regional de Taguatinga, Taguatinga, DF, Brasil Endereço para correspondência: Dr. Alexandre Dias Mançano Centro Médico Hospitalar Anchieta AE 8/10 Setor C Norte, 1º subsolo, Centro de Excelência Anchieta lojas 12 e 13 Taguatinga, DF, Brasil, 72115-700 E-mail: alex.manzano1@gmail.com Trabalho realizado na Radiologia Anchieta - Hospital Anchieta, Taguatinga, DF, Brasil. |
|
GN1© Copyright 2024 - All rights reserved to Colégio Brasileiro de Radiologia e Diagnóstico por Imagem
Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554
Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554