Radiologia Brasileira - Publicação Científica Oficial do Colégio Brasileiro de Radiologia
AMB - Associação Médica Brasileira CNA - Comissão Nacional de Acreditação
 Vol. 46 nº 3 - May / June of 2013
Vol. 46 nº 3 - May / June of 2013
|
ICONOGRAPHIC ESSAY
|
|
Rendu-Osler-Weber syndrome: what radiologists should know. Literature review and three cases report |
|
|
Autho(rs): Paulo Moraes Agnollitto1; André Rodrigues Façanha Barreto1; Raul Fernando Pinsetta Barbieri1; Jorge Elias Junior2; Valdair Francisco Muglia2 |
|
|
Keywords: Rendu-Osler-Weber; Hereditary hemorrhagic telangiectasia; Arteriovenous fistula; Epistaxis. |
|
|
Abstract: INTRODUCTION
Rendu-Osler-Weber syndrome or hereditary hemorrhagic telangiectasia (HHT) is an autosomal dominant disease with variable penetrance(1,2), involving multiple organs and systems, and characterized by the presence of angiodysplastic lesions which cause communication between arteries and veins without interposition of a capillary bed(3). Most common clinical manifestations include skin and mucosal telangiectasias and epistaxis, besides gastrointestinal, pulmonary and intracerebral bleeding(46). The reported prevalence of this disease is of 12/100,000 live neonates(7), but it may reach 1/3,500 to 1/5,000 in some regions(8). The first reports were published by Benjamin Guy Babington in 1865, John Wick-ham Legg in 1876, and Henri J. L. M. Rendu in 1886. Later, William Osler, Frederick Parkes Weber and Frederic M. Hanes described visceral manifestations and the hereditary nature of the disease(1). In the present study, the authors report three cases of this syndrome, highlighting the diagnostic criteria and imaging findings, with a literature review. CASE 1 A female, 57-year-old, brown-skinned patient complaining of repetition epistaxis and petechial lesions with perioral and periungueal distribution. At clinical examination, an ulcerated lesion was identified in the nasal septum. Such lesion was submitted to biopsy whose histopathological result revealed a chronic inflammatory process, with presence of round-shaped structures suggestive of the amastigote form of Leishmania sp. Chest radiography was performed as part of the investigation for the infectious condition (leishmaniasis), and detected the presence of a pulmonary nodule (Figure 1A). For further investigation, chest computed tomography (CT) was requested (Figure 1B) and demonstrated the vascular nature of the lesion. The hypothesis of HHT was raised and supplementary studies were requested abdominal and cranial CT and magnetic resonance imaging (MRI) (Figures 1 to 4). Epistaxis was treated with local tamponade. The follow-up was discontinued by the patient. 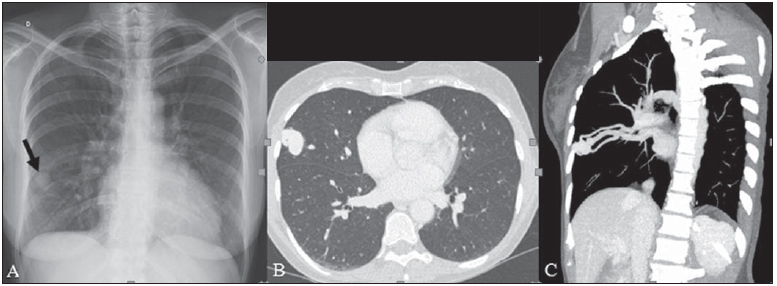 Figure 1. A: Posteroanterior chest radiography showing nodule with soft-tissue density in the right lung (arrow). B: Axial CT section, with maximum intensity projection (MIP) showing nodule with soft-tissue attenuation in the middle lobe of the right lung. C: Oblique MPR reconstruction demonstrating the vascular nature of the previously described nodule, with at least two pulmonary arterial branches supplying a malformative nidus and main pulmonary vein drainage (pulmonary arteriovenous malformation.  Figure 2. A: Contrast-enhanced, axial CT section demonstrating a lesion of vascular nature affecting the hepatic segment VII in association with dilatation of right hepatic vein (arrow). B: Axial MRI T2-weighted section identifying a probably portosystemic hepatic vascular malformation. C: Cross-sectional sections of color Doppler US demonstrating the vascular nature of the lesion. 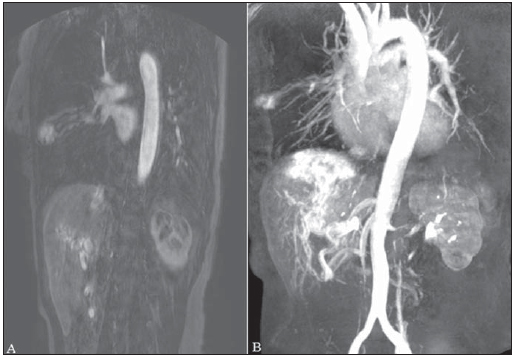 Figure 3. A: Gadolinium-enhanced, coronal MRI section demonstrating pulmonary nodular lesion of vascular nature, in agreement with CT. B: Contrast-enhanced abdominal MRI angiography demonstrating pulmonary and hepatic involvement by the disease. 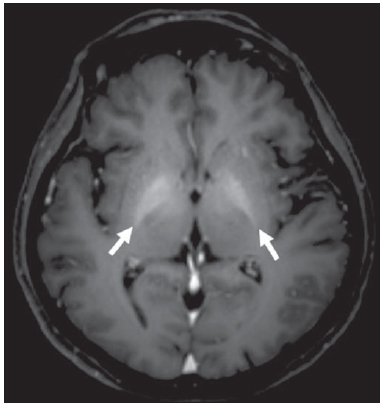 Figure 4. Axial brain MRI T1-weighted section demonstrating the presence of hypersignal in the globus pallidus (arrows), compatible with manganese deposition, confirming the presence of portosystemic shunt. CASE 2 A female, 43-year-old, white patient complaining of recurrent epistaxis for about 20 years, besides reddish lesions in her lips, tongue, hands and trunk. The patient underwent chest CT and abdominal CT angiography, which revealed typical findings of HHT (Figures 5 and 6). In no occasion treatment with tamponade was required and the patient is under follow-up for three years and has not presented any complication. This patient has also a daughter with recurrent epistaxis since the age of four, whose investigation is currently being initiated in the authors' institution, with no imaging study available until the present moment. 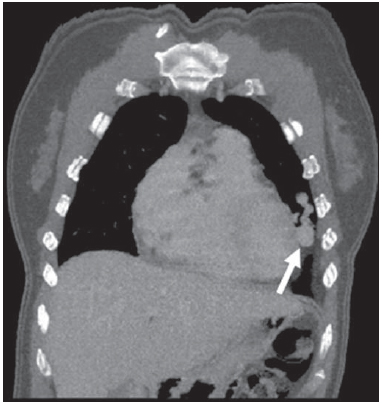 Figure 5. Coronal MPR reconstruction identifying a nodule with soft-tissue density associated with the presence of vessels in left lung inferior lingular segment. 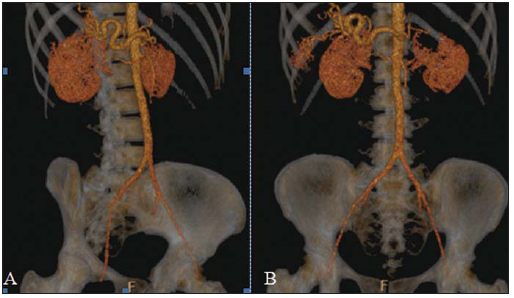 Figure 6. 3D reconstruction of CT angiography of the aorta demonstrating enlarged caliber in the whole extent of hepatic artery (A) and a cluster of serpiginous arterial vessels inside the liver parenchyma (B), compatible with intrahepatic fistula. CASE 3 A female, 36-year-old, white inpatient with fever was submitted to chest radiography which demonstrated the presence of pulmonary nodules. At physical examination, the patient presented digital hypocratism. Chest CT demonstrated the vascular nature of the lesions visualized at radiography (Figure 7). The infectious focus related to the fever was diagnosed as pyelonephritis not related to the alterations of HHT, satisfactorily treated with antibiotic therapy. During the diagnostic investigation, the positive family history for the disease could be revealed, since the patient had a relative with a past history of recurrent epistaxis. No specific treatment was required and the patient is under follow-up for six months, with neither complications nor symptoms related to the findings of the disease. 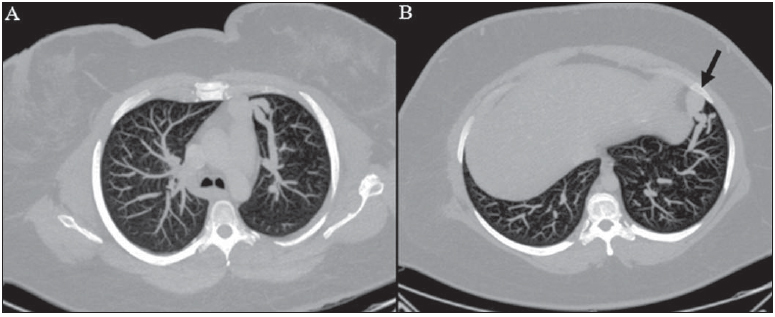 Figure 7. Axial chest CT with maximum intensity projection (MIP) algorithm demonstrating anomalous calibrous vessels in the pulmonary parenchyma (A), nodule in the anterior costophrenic sinus in continuity with dilated serpiginous vessels (B), characterizing pulmonary arteriovenous fistula. DISCUSSION The imaging evaluation of the vascular system has been discussed in a range of recent publication in the Brazilian radiological literature(920). HHT is diagnosed on the basis of the presence of three among four Curacao criteria(5,6), as follows 1) skin and mucosal telangiectasias; 2) recurrent spontaneous epistaxis; 3) visceral arteriovenous malformations (AVMs); 4) positive family history. HHT can be confirmed by means of the identification of the involved mutations with molecular biology techniques. All the cases reported in the present essay were diagnosed as HHT according the Curacao criteria. Two mutations are involved in the pathogenesis of this disease, namely: a) mutation of the endoglin gene located in the chromosome 9q that determines type 1 HHT, with marked pulmonary involvement; b) mutation of the activin gene located in the chromosome 12q that determines type 2 HHT with predominantly extrapulmonary involvement(4). The cases herein described were not genetically analyzed. HHT is characterized by progressive lifetime symptoms. Epistaxis is the first manifestation, with onset around the age of ten, and with increasingly severe episodes. Two of the cases presently reported had epistaxis as primary symptom, while the other had incidental detection of pulmonary lesions. The onset of telangiectasias is also progressive and the most common sites are the lips, tongue, palate, fingers and face. The main complications of the disease originate from pulmonary AVMs resulting in right-to-left shunting which may cause dyspnea, hemoptysis, paradoxical embolism and cerebral abscesses, which have not occurred in any of the cases presently reported. Hepatic vascular (arterioportal, arteriovenous and portosystemic) malformations may also cause right-to-left shunting and determine varied degrees of pulmonary hypertension, heart failure and hepatic encephalopathy. In a study with 35 children, Giordano et al.(21) have found nasal telangiectasias in 68%, skin and mucosal telangiectasias (fingers, lips, oral cavity) in 79%, pulmonary AVMs in 53%, hepatic AVMs in 47% and brain MAVs and/or ischemic brain alterations secondary to pulmonary AVMs in 12% of the patients. Brain AVMs may cause migraine and intraparenchymal and/or subarachnoid hemorrhage. None of the cases described in the present study presented neurological symptoms. Another neuroimaging finding that may aid in the diagnosis of HHT already described in association with congenital portosystemic shunts and clearly seen in the case 1 , is the presence of hypersignal on T1-weighted images of the basal nuclei, particularly of the globus pallidus, which indicates accumulation of manganese in the central nervous system. Such finding is nonspecific and may be associated with conditions such as parenteral nutrition, chronic hepatopathy with or without encephalopathy, acquired or congenital portosystemic shunts, and chronic dialytic renal failure with increased manganese serum levels(22,23). Differential diagnoses include capillary skin malformations with visceral AVMs (involving soft parts, brain or bones) caused by RASA1 mutation; Adams-Oliver syndrome (with skin telangiectasias, pulmonary AVMs, and limb and scalp defects); or vascular retino-encephalic malformation (Wyburn-Mason syndrome), which may falsely meet the Curacao criteria for HHT. The treatment for HHT includes measures for management of epistaxis (local tamponade and laser); management of iron deficiency anemia with iron supplementation and transfusions, as necessary; management of pulmonary, cerebral and hepatic AVMs with careful embolization or, alternatively, surgical resection, radiotherapy and liver transplantation, respectively(1). The two cases with epistaxis were treated with local tamponade, while one of the patients was submitted to endovascular embolization of pulmonary AVM because of hemoptysis. The most frequent situations related to this rare syndrome in the radiologist's daily routine include: a) incidental finding of a visceral AVM in a patient without previous diagnosis of HHT; b) examination targeted to the investigation or management of visceral alterations in patients already diagnosed with HHT. In the present cases, it is observed that the diagnosis was achieved mainly by the detection of epistaxis and hemoptysis, while in one case a vascular pulmonary lesion was incidentally found. Therefore, the authors conclude that, in the presence of a finding suggestive of visceral AVM, it is important that the radiologist has the HHT diagnostic criteria in mind, either asking the requesting physician or directly asking the patient at the moment of the procedure. In the cases where patients with recurrent epistaxis or hemoptysis submitted to chest or abdominal imaging, attention should be paid to the presence of lesions originating from vascular malformations. Once the diagnosis of HHT is established in adult patients, screening for vascular abdominal (particularly hepatic) and pulmonary lesions should be performed in the children of such patients' families, with abdominal ultrasonography, plain radiography and, eventually, chest CT(9). REFERENCES 1. Cottin V, Dupuis-Girod S, Lesca G, et al. Pulmonary vascular manifestations of hereditary hemorrhagic telangiectasia (rendu-osler disease). Respiration. 2007;74:36178. 2. Sabbà C, Pasculli G, Cirulli A, et al. Rendu-Osler-Weber disease: experience with 56 patients. Ann Ital Med Int. 2002;17:1739. 3. Memeo M, Scardapane A, De Blasi R, et al. Diagnostic imaging in the study of visceral involvement of hereditary haemorrhagic telangiectasia. Radiol Med. 2008;113:54766. 4. Carette MF, Nedelcu C, Tassart M, et al. Imaging of hereditary hemorrhagic telangiectasia. Cardiovasc Intervent Radiol. 2009;32:74557. 5. Sadick H, Sadick M, Götte K, et al. Hereditary hemorrhagic telangiectasia: an update on clinical manifestations and diagnostic measures. Wien Klin Wochenschr. 2006;118:7280. 6. Siddiki H, Doherty MG, Fletcher JG, et al. Abdominal findings in hereditary hemorrhagic telangiectasia: pictorial essay on 2D and 3D findings with isotropic multiphase CT. Radiographics. 2008;28:17184. 7. Buscarini E, Buscarini L, Civardi G, et al. Hepatic vascular malformations in hereditary hemorrhagic telangiectasia: imaging findings. AJR Am J Roentgenol. 1994;163:110510. 8. Sabba C, Pasculli G, Cirulli A, et al. Hereditary hemorrhagic teleangiectasia (Rendu-Osler-Weber disease). Minerva Cardioangiol. 2002;50:22138. 9. Bezerra KB, Bacelar Jr EA, Pereira NCS, et al. Gastric arteriovenous malformation: treatment by embolization. Radiol Bras. 2012;45:1268. 10. Monteiro HF, Torres PPTS, Edelhoff VNG, et al. Nutcracker syndrome [which is your diagnosis?]. Radiol Bras. 2012;45:xiiixiv. 11. Brandão AHF, Barbosa AS, Lopes APBM, et al. Dopplerfluxometry of ophthalmic arteries and assessment of endothelial function in early and late preeclampsia. Radiol Bras. 2012;45:203. 12. Novero ER, Metzger PB, Obregon J, et al. Endovascular treatment of thoracic aortic diseases: a single center result analysis. Radiol Bras. 2012;45:2518. 13. Novero ER, Metzger PB, Angelieri FMR, et al. Endovascular repair of abdominal aortic aneurysm: a single-center results analysis. Radiol Bras. 2012;45:16. 14. Farret Neto A, Faria EDB, Pantaleo E, et al. Anterior tibial artery recanalization through dorsalis pedis artery: a case report. Radiol Bras. 2012;45:3024. 15. Guimarães Filho A, Carneiro Neto LA, Palheta MS, et al. Caroli's disease complicated with liver abscess: case report. Radiol Bras. 2012;45:3624. 16. Iyeyasu JN, Silva Júnior NA, Bertanha R, et al. Carotid-cavernous fistula [which is your diagnosis?]. Radiol Bras. 2012;45:xiiixiv. 17. Iared W, D'Ippolito G. Doppler flowmetry in hepatic steatosis. Radiol Bras. 2011;44(1):v. 18. Ceratti S, Okano FM, Pontes ABG, et al. Ultrasound-guided foam sclerotherapy in the treatment of chronic venous insufficiency. Radiol Bras. 2011;44:16771. 19. Wajnberg E, Rodrigues G, Abud DG. Use of drug-eluting stents for the treatment of vertebral artery stenosis. Radiol Bras. 2011;44:3438. 20. Bastos AL, Brito ILA. Pulmonary artery aneurysms in Behçet's disease: case report. Radiol Bras. 2011;44:3968. 21. Giordano P, Nigro A, Lenato GM, et al. Screening for children from families with Rendu-Osler-Weber disease: from geneticist to clinician. J Thromb Haemost. 2006;4:123745. 22. da Silva CJ, da Rocha AJ, Jeronymo S, et al. A preliminary study revealing a new association in patients undergoing maintenance hemodialysis: manganism symptoms and T1 hyperintense changes in the basal ganglia. AJNR Am J Neuroradiol. 2007;28:14749. 23. Rovira A, Alonso J, Cordoba J. MR imaging findings in hepatic encephalopathy. AJNR Am J Neuroradiol. 2008;29:161221. 1. MDs, Residents of Radiology and Imaging Diagnosis, Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo (HC-FMRPUSP), Ribeirão Preto, SP, Brazil. 2. Private Docents, Docents of the Center of Imaging Sciences and Medical Physics, Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo (HC-FMRPUSP), Ribeirão Preto, SP, Brazil. * Study developed at Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo (HC-FMRPUSP), Ribeirão Preto, SP, Brazil. Mailing Address: Dr. Paulo Moraes Agnollitto Avenida Caramuru, 2300, República Ribeirão Preto, SP, Brazil, 14030-000 E-mail: agno53@gmail.com Received August 28, 2012. Accepted after revision February 15, 2013. |
|
Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554