Radiologia Brasileira - Publicação Científica Oficial do Colégio Brasileiro de Radiologia
AMB - Associação Médica Brasileira CNA - Comissão Nacional de Acreditação
 Vol. 45 nº 3 - Maio / Jun. of 2012
Vol. 45 nº 3 - Maio / Jun. of 2012
|
QUAL O SEU DIAGNÓSTICO?
|
|
|
|
|
Autho(rs): Alair Augusto Sarmet Moreira Damas dos Santos1; Camila Specht Silva Menezes2; Wolney de Castro Figueiredo3 |
|
|
Paciente do sexo masculino, pardo, 50 anos de idade, com hipogonadismo hipogonadotrófico e anosmia, foi atendido no Serviço de Endocrinologia do Hospital Universitário Antonio Pedro, onde realizou exame de ressonância magnética de sela turca e crânio (Figuras 1 e 2).
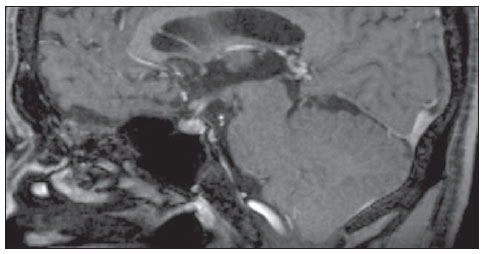 Figura 1. Ressonância magnética da sela turca no plano sagital ponderada em T1 pós-contraste. 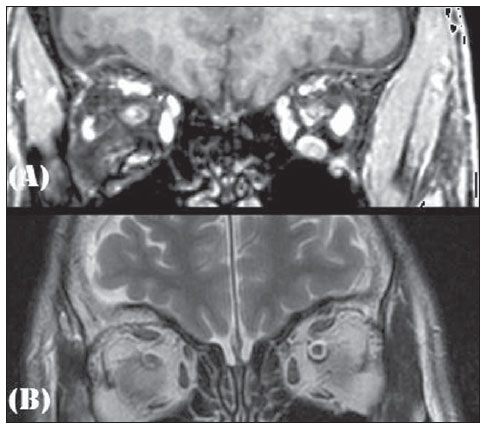 Figura 2. Ressonância magnética em corte coronal T1 (A) e T2 (B). Em 1982, quando tinha 21 anos, procurou Serviço Público de Endocrinologia e na ocasião realizou exames complementares, com os seguintes valores: LH basal = 10,0 mUI/ml (v.r. pré-pu- Trabalho realizado na Faculdade de Medicina da Universidade Federal Fluminense (UFF), Niterói, RJ, Brasil. 1. Doutor, Professor Associado de Radiologia da Faculdade de Medicina da Universidade Federal Fluminense (UFF), Chefe do Serviço de Radiologia do Hospital Universitário Antonio Pedro (HUAP), Niterói, RJ, Brasil. 2. Interna da Faculdade de Medicina da Universidade Federal Fluminense (UFF), Niterói, RJ, Brasil. 3. Médico, Professor beral: 2,012,0 mUI/ml); FSH basal = 2,0 mUI/ml (v.r. pré-puberal: 2,012,0 mUI/ml); testosterona = 38,0 ng/dl (v.r.: 2451600 ng/dl); prolactina = 10 ng/ml (v.r.: 025 ng/ml); teste provocativo com LHRH evidenciou deficiência isolada de LH; teste provocativo com clomifeno com ausência de resposta; cariótipo 46,XY; idade óssea estimada em 15 anos; azoospermia; radiografia de crânio normal; tomografia de sela turca normal; ultrassonografia abdominal total normal. Descrição das imagens Figura 1. Ressonância magnética da sela turca no plano sagital ponderada em T1 pós-contraste evidencia hipófise e haste hipofisária normais. Figura 2. Ressonância magnética em corte coronal T1 (A) e T2 (B) evidencia ausência de bulbo olfatório. Diagnóstico: Síndrome de Kallmann. COMENTÁRIOS A síndrome de Kallmann é uma rara neuropatia que cursa com hipogonadismo hipogonadotrófico associado com anosmia ou hiposmia. Sua incidência é de 1:10.000 homens e 1:50.000 mulheres. Neste desafio diagnóstico é apresentado um paciente com quadro clínico de síndrome de Kallmann avaliado por ressonância magnética, que revelou ausência de sulcos e bulbos olfatórios e hipófise sem alterações, que são os achados clássicos que corroboram o diagnóstico desta doença(1). A síndrome de Kallmann decorre de um defeito embrionário na migração dos neurônios do bulbo olfatório e os neurônios secretores de GnRH, que se originam no epitélio nasal e migram através da placa cribriforme em direção ao bulbo olfatório e área pré-optica do hipotálamo, respectivamente. A desorientação desta migração provoca alteração da olfação e na secreção do GnRH, estabelecendo a díade da síndrome de Kallmann: hipogonadismo hipogonadotrófico e anosmia(13). O diagnóstico é essencialmente clínico, com confirmação bioquímica de baixos níveis de esteróides sexuais, níveis baixos ou normais de LH e FSH e imagem associada a função normal da hipófise. Nas últimas décadas, grandes avanços nas pesquisas em neuroimagem permitiram um grande passo na elucidação da síndrome de Kallmann. O advento da ressonância magnética representou importante elemento para o diagnóstico, pois permite correlacionar clinicamente a anosmia ou hiposmia com alterações de agenesia ou hipoplasia de bulbo e estrias olfatórias(1). Na ressonância magnética, os bulbos olfatórios são mais bem visualizados no plano coronal, como estruturas junto da placa cribriforme, e os sulcos olfatórios são vistos entre o giro reto e o giro orbital medial( 2). No exame do paciente em questão há os achados típicos da síndrome. Esta imagem característica, associada à clínica, permite determinar o diagnóstico definitivo da síndrome de Kallmann, assim como o diagnóstico diferencial com hipogonadismo hipogonadotrófico idiopático(1,2,4,5). Nos casos em que a apresentação não é clássica ou em crianças pré-puberais, nas quais a clínica ainda não se estabeleceu integralmente, a neuroimagem na ressonância magnética, associado ao estudo genético, é ponto chave na presunção da síndrome de Kallmann(1). É vital a correta realização deste exame de imagem, com especial avaliação do plano coronal, uma vez que as alterações são sutis e podem passar despercebidas, devendo o médico radiologista buscar a correlação clínico-laboratorial com os achados no exame. REFERÊNCIAS 1. Pritteloud N, Crowley WF Jr. Congenital gonadotropin- releasing hormone deficiency (idiopathic hypogonadotropic hypogonadism). UpToDate 2011. [acessado em 21 de julho de 2011]. Disponível em: uptodate.com 2. Madan R, Sawlani V, Gupta S, et al. MRI findings in Kallmann syndrome. Neurol India. 2004;52:5013. 3. Franco B, Guiole S, Pragliola A, et al. A gene deleted in Kallmann's syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature. 1991;353:52936. 4. Abolmaali ND, Hietschold V, Vogl TJ, et al. MR evaluation in patients with isolated anosmia since birth or early childhood. AJNR Am J Neuroradiol. 2002;23:15764. 5. Truwit CL, Barkovich AJ, Grumbach MM, et al. MR imaging of Kallmann syndrome, a genetic disorder of neuronal migration affecting the olfactory and genital systems. AJNR Am J Neuroradiol. 1993;14:82738. 1. Doutor, Professor Associado de Radiologia da Faculdade de Medicina da Universidade Federal Fluminense (UFF), Chefe do Serviço de Radiologia do Hospital Universitário Antonio Pedro (HUAP), Niterói, RJ, Brasil. 2. Interna da Faculdade de Medicina da Universidade Federal Fluminense (UFF), Niterói, RJ, Brasil. 3. Médico, Professor Adjunto de Endocrinologia da Faculdade de Medicina da Universidade Federal Fluminense (UFF), Niterói, RJ, Brasil. Endereço para correspondência: Camila Specht Silva Menezes Rua Bulhões de Carvalho, 378, ap. 1001, Copacabana Rio de Janeiro, RJ, Brasil, 22081-000 E-mail: specht.camila@hotmail.com Trabalho realizado na Faculdade de Medicina da Universidade Federal Fluminense (UFF), Niterói, RJ, Brasil. |
|
GN1© Copyright 2024 - All rights reserved to Colégio Brasileiro de Radiologia e Diagnóstico por Imagem
Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554
Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554