Radiologia Brasileira - Publicação Científica Oficial do Colégio Brasileiro de Radiologia
AMB - Associação Médica Brasileira CNA - Comissão Nacional de Acreditação
 Vol. 45 nº 3 - May / June of 2012
Vol. 45 nº 3 - May / June of 2012
|
WHICH IS YOUR DIAGNOSIS?
|
|
Which is your diagnosis? |
|
|
Autho(rs): Alair Augusto Sarmet Moreira Damas dos Santos1; Camila Specht Silva Menezes2; Wolney de Castro Figueiredo3 |
|
|
A male, 50-year-old with hypogonadotrophic hypogonadism and anosmia was assisted in the Unit of Endocrinology of Hospital Universitário Antonio Pedro, being submitted to magnetic resonance imaging of the head and sella turcica (Figures 1 and 2).
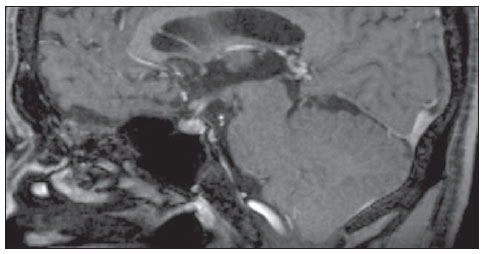 Figure 1. Magnetic resonance imaging of sella turcica contrast-enhanced, sagittal, T1-weighted image. 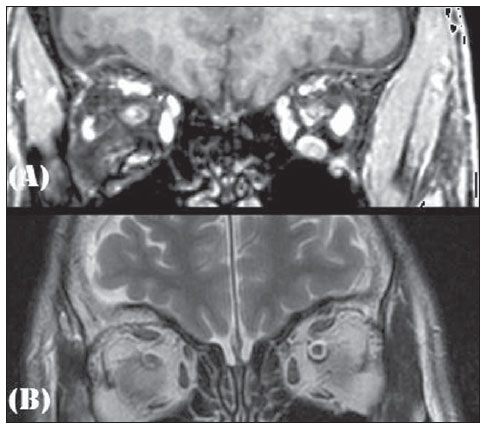 Figure 2. Magnetic resonance imaging coronal, T1-weighted (A) and T2-weighted (B) images. In 1982, the patient who, at that time was 21-year-old, sought the Public Endocrinology Service and was submitted to laboratory tests with the following results: basal LH level = 10.0 mUI/ ml (prepubertal reference values: 2.0-12.0 mUI/ml); basal FSH level = 2.0 mUI/ml (prepubertal reference values: 2.0-12.0 mUI/ ml); testosterone = 38.0 ng/dl (reference values: 245-1600 ng/dl); prolactin = 10 ng/ml (reference values: 0-25 ng/ml); LHRH stimulation test demonstrated isolated LH deficiency; clomiphene stimulation test with absence of response; 46,XY karyotype; besides estimated bone age corresponding to 15 years; azoospermia; normal cranial radiography; normal computed tomography of the sella turcica; and normal whole abdomen ultrasonography. Images description Figure 1. Magnetic resonance imaging of sella turcica. Contrast-enhanced, sagittal, T1-weighted image demonstrating normal hypophysis and hypophyseal stalk. Figure 2. Magnetic resonance imaging. Coronal, T1-weighted (A) and T2-weighted (B) images demonstrate absence of olfactory bulbs. Diagnosis: Kallmann syndrome. COMMENTS Kallmann syndrome is a rare neuropathy that courses with hypogonadotrophic hypogonadism associated either with anosmia or hyposmia. The incidence of such a condition corresponds to 1:10,000 men and 1:50,000 women. The present diagnostic challenge presents a patient with clinical signs of Kallmann syndrome who was assessed by magnetic resonance imaging, demonstrating absence of olfactory bulbs and sulci with unaltered hypophysis which constitute the typical findings corroborating the diagnosis of this disease(1). Kallmann syndrome originates from an embryonic defect in the neuronal migration into the olfactory bulb and the GnRH neurons originating from the nasal epithelium and migrate through the cribriform plate towards the olfactory bulb and the preoptic area of the hypothalamus, respectively. Such a disoriented migration causes alteration in the olfaction and in GnRH secretion, constituting the couple of characteristics of the Kallmann syndrome: hypogonadotrophic hypogonadism and anosmia(1-3). The syndrome diagnosis is essentially clinical, with biochemical confirmation of low serum levels of sex steroids, low or normal levels of LH and FSH, and image associated with a normal hypophyseal function. In the last decades, huge developments in the research of neuroimaging has allowed a great progress in the elucidation of the Kallmann syndrome. The arrival of magnetic resonance imaging has represented an important element for the diagnosis of this syndrome, for allowing the clinical correlation between anosmia or hyposmia and agenesia or hypoplasia of olfactory bulb and striae(1). At magnetic resonance imaging, the olfactory bulbs are better visualized in the coronal plane, as structures adjacent to the cribriform plate, and the olfactory sulci are seen between the straight gyrus and the medial orbital gyrus(2). The patient assessment reveals typical findings of this syndrome. Such characteristic image, in association with clinical findings, determines the definite diagnosis of Kallmann syndrome, as well as the differential diagnosis with idiopathic hypogonadotrophic hypogonadism(1,2,4,5). In cases of non-classical presentation or disorder in prepubertal children in which the clinical findings are still to be integrally established, magnetic resonance neuroimaging, in association with genetic investigation, is a key method in Kallmann syndrome presumption(1). A correct imaging, especially in the coronal plane, is of paramount importance, considering the subtlety of alterations which may go unperceived, thus the radiologist should establish a clinical/laboratory correlation with imaging findings. REFERENCES 1. Pritteloud N, Crowley WF Jr. Congenital gonadotropin- releasing hormone deficiency (idiopathic hypogonadotropic hypogonadism). UpToDate 2011. [acessado em 21 de julho de 2011]. Disponível em: uptodate.com 2. Madan R, Sawlani V, Gupta S, et al. MRI findings in Kallmann syndrome. Neurol India. 2004;52:5013. 3. Franco B, Guiole S, Pragliola A, et al. A gene deleted in Kallmann's syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature. 1991;353:52936. 4. Abolmaali ND, Hietschold V, Vogl TJ, et al. MR evaluation in patients with isolated anosmia since birth or early childhood. AJNR Am J Neuroradiol. 2002;23:15764. 5. Truwit CL, Barkovich AJ, Grumbach MM, et al. MR imaging of Kallmann syndrome, a genetic disorder of neuronal migration affecting the olfactory and genital systems. AJNR Am J Neuroradiol. 1993;14:82738. 1. PhD, Associate Professor of Radiology, School of Medicine Universidade Federal Fluminense (UFF), Head of Service of Radiology at Hospital Universitário Antonio Pedro (HUAP), Niterói, RJ, Brazil. 2. Graduate Student of Medicine, School of Medicine Universidade Federal Fluminense (UFF), Niterói, RJ, Brazil. 3. MD, Associate Professor of Endocrinology at School of Medicine Universidade Federal Fluminense (UFF), Niterói, RJ, Brazil. Mailing Address: Camila Specht Silva Menezes Rua Bulhões de Carvalho, 378, ap. 1001, Copacabana Rio de Janeiro, RJ, Brazil, 22081-000 E-mail: specht.camila@hotmail.com Study developed at School of Medicine Universidade Federal Fluminense (UFF), Niterói, RJ, Brazil. |
|
GN1© Copyright 2024 - All rights reserved to Colégio Brasileiro de Radiologia e Diagnóstico por Imagem
Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554
Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554