Radiologia Brasileira - Publicação Científica Oficial do Colégio Brasileiro de Radiologia
AMB - Associação Médica Brasileira CNA - Comissão Nacional de Acreditação
 Vol. 44 nº 3 - Maio / Jun. of 2011
Vol. 44 nº 3 - Maio / Jun. of 2011
|
RELATO DE CASO
|
|
|
|
|
Autho(rs): Rodrigo Manfroi Gutsche1; Lucia Antunes Chagas2; Rodolfo Leal2; André Lima da Cunha3; Maria Célia Resende Djahjah4 |
|
|
Descritores: Ductos paramesonéfricos; Anormalidades. |
|
|
Resumo: INTRODUÇÃO
A síndrome de Mayer-Rokitansky-Kuster-Hauser é uma condição incomum, com incidência de um para cada 4.0005.000 nascimentos de meninas(1,2), sendo a segunda causa mais frequente de amenorreia primária, após as disgenesias gonadais(3). É uma forma de agenesia mülleriana caracterizada por atresia vaginal, anomalias uterinas e tubárias, que podem incluir ausência ou hipoplasia. As pacientes apresentam cariótipo 46,XX e caracteres sexuais secundários normais, uma vez que os ovários estão presentes e funcionantes, porém não há menstruação(4). A síndrome se apresenta em três formas, classificadas segundo o acometimento de estruturas além do aparelho reprodutor. A síndrome típica, tipo I, é representada por alterações restritas ao sistema reprodutor. A segunda, tipo II, é uma síndrome atípica, na qual estão presentes assimetria no remanescente uterino e anomalia das tubas uterinas. Esta forma pode estar associada a doença ovariana, alterações renais, ósseas e otológicas congênitas. Um terceiro tipo, denominado MURCS, envolve hipoplasia ou aplasia uterovaginal, malformações renais, ósseas e cardíacas(13). As malformações renais que podem ser encontradas são agenesia unilateral, rim em ferradura, hipoplasia renal, rins ectópicos e hidronefrose. As malformações ósseas ocorrem principalmente nas vértebras, sendo mais comuns a fusão de vértebras, principalmente cervicais, síndrome de Klippel-Feil e escoliose. As alterações cardíacas e digitais, como sindactilia e polidactilia, são mais raras que as anteriores(1). A etiologia permanece desconhecida, porém o aumentado número de casos em grupos familiares ressalta a hipótese de uma causa genética(1). Neste relato apresentamos o caso de uma paciente de 16 anos de idade, com amenorreia primária e caracteres sexuais secundários normais, submetida a investigação clinicorradiológica e tratamento cirúrgico. RELATO DO CASO Paciente do sexo feminino, 16 anos de idade, com amenorreia primária, sem outras afecções clínicas. Ao exame clínico constatou-se desenvolvimento dos caracteres sexuais secundários compatível com a idade cronológica. Ao exame ginecológico observou-se uretra em fenda com bordas elevadas. Não foi realizado exame ginecológico com espéculo. A ultrassonografia transpélvica não evidenciou imagem uterina e ovários em seus sítios habituais, no entanto, foi inconclusiva por causa de dificuldades técnicas. À ressonância magnética, o útero não foi visualizado e os ovários possuíam volume, sinal e localização sem anormalidades. O canal vaginal também não foi visualizado por este método (Figura 1). 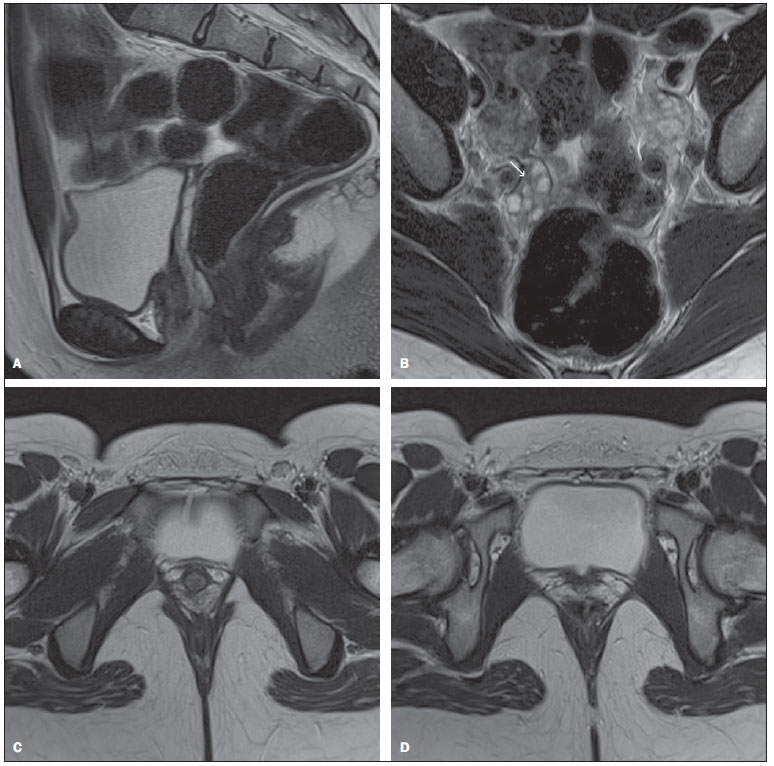 Figura 1. A: Imagem no plano sagital T2, sem supressão de gordura: observar loja uterina vazia. B: Imagem ponderada em T2, sem supressão de gordura, no plano axial: observar ovários com forma e volume normais, em sítio habitual, o direito apresentando folículos (seta). C: Imagem ponderada em T2, no plano axial: observar ausência da vagina. D: Imagem ponderada em T2, no plano axial: observar a ausência do útero. A avaliação genética revelou cariótipo 46,XX, determinando, desse modo, o diagnóstico de síndrome de Mayer-Rokitansky-Kuster-Hauser. A paciente foi submetida a correção cirúrgica da vagina (neovaginoplastia). DISCUSSÃO A apresentação clínica típica é amenorreia primária, acompanhada ou não de cólicas cíclicas, em adolescente com caracteres sexuais secundários compatíveis com a idade, sem sinais de virilização. O exame ginecológico pode detectar ausência do canal vaginal ou encurtamento da vagina(1,2,58). A realização de exames de imagem, como a ultrassonografia e a ressonância magnética, associados ou não à laparoscopia, é necessária para que se possam determinar as características anatômicas da síndrome. A ultrassonografia é o primeiro exame a ser solicitado. Este exame pode revelar a ausência do útero entre a bexiga e o reto(1,5,9). A lâmina vestigial, quando encontrada no sítio habitual do útero, pode ser confundida com este. Ainda podem ser observadas anomalias renais quando há síndrome do tipo II(1). A ressonância magnética é o método de imagem que apresenta maior sensibilidade e especificidade na avaliação da síndrome, não apenas por permitir a realização de imagens multiplanares, mas também por possibilitar a obtenção de sequências com saturação de gordura. Permite boa definição de alterações anatômicas como a agenesia uterina, além de avaliação do ovário, vagina e anomalias associadas(1,4,5,9). A laparoscopia está indicada apenas quando a avaliação pelos dois métodos anteriores for insatisfatória e for possível, por este ato, traçar uma conduta terapêutica. Após o diagnóstico de síndrome de Mayer-Rokitansky-Kuster-Hauser, deve-se fazer uma investigação clínica para a identificação de possíveis malformações associadas(1,4). O diagnóstico final é a associação dos achados nesses métodos com o cariótipo. O diagnóstico diferencial deve ser feito com outras situações em que a paciente apresenta amenorreia primária e caracteres sexuais secundários desenvolvidos, como ausência congênita de útero e vagina, atresia vaginal isolada com síndrome da insensibilidade androgênica e septo vaginal transverso com hímen imperfurado(1). A síndrome de Mayer-Rokitansky-Kuster-Hauser, pelas alterações anatômicas que a caracteriza, gera ansiedade e consequências psicológicas e na qualidade de vida das pacientes, devendo por isso ter uma abordagem multidisciplinar(5,6). O tratamento anatômico indicado é a criação de uma neovagina, de modo cirúrgico ou não, o que pode permitir a essas pacientes uma vida sexual normal(16). Quando a via cirúrgica é escolhida, remanescentes uterinos podem ser retirados, com o intuito de evitar endometriose futura(1). Às pacientes que desejam ter filhos deve ser encorajada a adoção e apresentada a possibilidade de ter filhos biológicos por meio da técnica de reprodução assistida, uma vez que, por possuírem ovários funcionantes, essas mulheres produzem óvulos normais(10). Mesmo com os recentes avanços no manejo desta síndrome, o seu diagnóstico traz significativo impacto psicológico e na qualidade de vida das pacientes, em razão da impossibilidade de menstruar e de engravidar. O estresse causado pelo diagnóstico pode ser aliviado pelos tratamentos cirúrgico ou não cirúrgico, pela passagem do tempo, pelo aconselhamento, pelo suporte de familiares e por grupos de ajuda(10). REFERÊNCIAS 1. Morcel K, Camborieux L. Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome. Orphanet J Rare Dis. 2007;2:13. 2. Sultan C, Biason-Lauber A, Philibert P. Mayer-Rokitansky-Kuster-Hauser syndrome: recent clinical and genetic findings. Gynecol Endocrinol. 2009;25:811. 3. Fedele L, Bianchi S, Tozzi L, et al. A new laparoscopic procedure for creation of a neovagina in Mayer-Rokitansky-Kuster-Hauser syndrome. Fertil Steril. 1996;66:854. 4. Sem KK, Kapoor A. Mayer-Rokitansky-Kuster-Hauser syndrome. Ind J Radiol Imag. 2006;16:8057. 5. Junqueira BLP, Allen LM, Spitzer RF, et al. Müllerian duct anomalies and mimics in children and adolescents: correlative intraoperative assessment with clinical imaging. Radiographics. 2009;29:1085103. 6. Muelle GC, Hussain HK, Smith YR. Müllerian duct anomalies: comparison of MRI diagnosis and clinical diagnosis. AJR Am J Roentgenol. 2007;189:1294302. 7. Troiano RN, McCarthy SM. Müllerian duct anomalies: imaging and clinical issues. Radiology. 2004;233:1934. 8. Stainkeler FA, Woodfield CA, Hillstrom MM. Female infertility: a systematic approach to radiologic imaging and diagnosis. Radiographics. 2009;29:135370. 9. Strübbe EH, Willemsen WNP, Lemmens JAM, et al. Mayer-Rokitansky-Kuster-Hauser syndrome: distinction between two forms based on excretory urographic, sonographic, and laparoscopic findings. AJR Am J Roentgenol. 1993;160:3314. 10. Bean EJ, Mazur T, Robinson AD. Mayer-Rokitansky-Kuster-Hauser: sexuality, psychological effects, and quality of life. J Pediatr Adolesc Gynecol. 2009;22:33946. 1. Médico Residente de Radiologia do Hospital Universitário Clementino Fraga Filho da Universidade Federal do Rio de Janeiro (HUCFF-UFRJ), Rio de Janeiro, RJ, Brasil. 2. Acadêmicos de Medicina da Universidade Federal do Rio de Janeiro (UFRJ), Rio de Janeiro, RJ, Brasil. 3. Cirurgião Pediátrico do Instituto de Puericultura e Pediatria Martagão Gesteira (IPPMG), Rio de Janeiro, RJ, Brasil. 4. Médica do Serviço de Radiologia do Hospital Universitário Clementino Fraga Filho da Universidade Federal do Rio de Janeiro (HUCFF-UFRJ), Rio de Janeiro, RJ, Brasil. Endereço para correspondência: Dr. Rodrigo Manfroi Gutsche Rua Conde de Bonfim, 850, ap. 803, Bloco 1, Tijuca Rio de Janeiro, RJ, Brasil, 20530-002 E-mail: rgmanfroi@yahoo.com.br Recebido para publicação em 4/10/2010. Aceito, após revisão, em 11/2/2011. Trabalho realizado no Hospital Universitário Clementino Fraga Filho da Universidade Federal do Rio de Janeiro (HUCFF-UFRJ), Rio de Janeiro, RJ, Brasil. |
|
Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554