Radiologia Brasileira - Publicação Científica Oficial do Colégio Brasileiro de Radiologia
AMB - Associação Médica Brasileira CNA - Comissão Nacional de Acreditação
 Vol. 43 nº 4 - Jul. / Ago. of 2010
Vol. 43 nº 4 - Jul. / Ago. of 2010
|
RELATO DE CASO
|
|
|
|
|
Autho(rs): Lívia Teresa Moreira Rios1; Marília da Glória Martins2; Vanda Maria Ferreira Simões3; Marynéa do Vale Nunes4; Patrícia Franco Marques5; Silvia Helena Cavalcante de Souza Godoy6 |
|
|
Descritores: Acalvaria primária; Defeito do crânio; Malformação congênita. |
|
|
Resumo: INTRODUÇÃO
Acalvaria é uma malformação congênita rara, de patogênese desconhecida, na qual os ossos da abóbada craniana, a duramáter e a musculatura associada estão ausentes, com base de crânio e ossos faciais normais. O tecido cerebral costuma estar preservado, embora, em alguns casos, possa estar anormalmente desenvolvido(1-4). Tem sido descrita como anomalia fatal, com raros relatos na literatura(5). Este relato descreve os achados de imagem da acalvaria primária, marcada pela ausência de ossos da abóbada craniana. RELATO DO CASO Primigesta de 15 anos de idade, foi encaminhada ao pré-natal especializado de nossa instituição com 38 semanas e 3 dias de gestação, portando exame ultrassonográfico com relato de hidrocefalia fetal unilateral diagnosticada com 31 semanas. Não havia história materna de medicação teratogênica, infecção recente, diabetes mellitus, hipertensão ou exposição a drogas na gestação atual. Foi indicada cesariana. Neonato do sexo feminino nasceu a termo pesando 2.815 gramas. Ao exame físico, a face da recém-nascida aparentava normalidade, com pele presente na região frontal. Destacavam-se proeminência dos hemisférios cerebrais e grande defeito na abóbada craniana. O defeito caracterizava-se pela ausência parcial do couro cabeludo e de ossos da abóbada craniana, com fina camada membranosa recobrindo o tecido cerebral, a partir da qual se observava extravasamento de líquido cefalorraquidiano compatível com a pia-máter (Figura 1). Não se palpavam os ossos frontais e parietais. Foi encaminhada à UTI-Neonatal, onde se prosseguiu investigação. 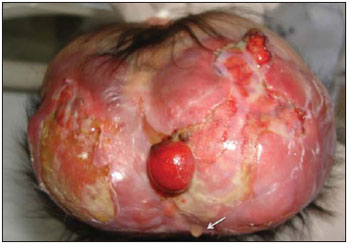 Figura 1. Exame físico evidencia ausência parcial do couro cabeludo e de ossos da abóbada craniana. Hemisférios cerebrais recobertos por fina membrana correspondente à pia-máter, de onde extravasa líquor (seta). Observam-se áreas de menor resistência com exposição do tecido cerebral. Uma radiografia do crânio, logo após o nascimento, revelou ausência dos ossos frontais, parietais e temporais, com faciais e occipital presentes (Figura 2). Os achados foram confirmados por tomografia computadorizada, que também demonstrou dilatação ventricular unilateral (Figura 3). Exame ultrassonográfico abdominal e ecocardiograma não revelaram anormalidades. 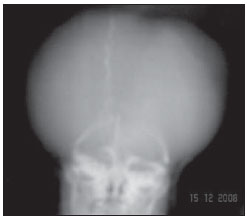 Figura 2. Radiografia do crânio logo após o nascimento demonstra ausência de ossos da abóbada craniana. Ossos faciais normais. 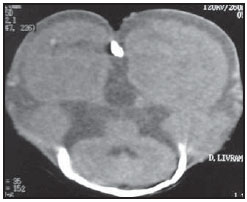 Figura 3. Tomografia do crânio realizada no segundo dia de vida confirmando achados clínicos e radiográficos. O osso occipital está preservado. O cérebro permaneceu recoberto pela pia-máter por cinco dias. No sexto dia de vida, em função de seu crescimento, o tecido cerebral começou a romper a fina membrana e ficou completamente exposto no décimo dia de vida. Uma tentativa de fechamento do defeito ósseo foi realizada com pericárdio bovino no 12º dia de vida. A reconstrução evoluiu com quadro infeccioso local e deiscência de parte da sutura. Após dois meses de vida, já havia sinais clínicos de comprometimento neurológico hipóxico-isquêmico. Exame ultrassonográfico transfontanelar, após quatro meses de vida, revelou áreas císticas encefálicas decorrentes de lesão vascular hipóxico-isquêmica (Figura 4). A recém-nascida evoluiu para o óbito no quinto mês de vida, por complicações respiratórias e infecciosas. 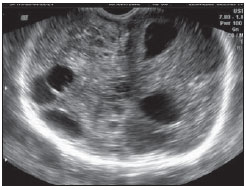 Figura 4. Ultrassonografia transfontanelar de controle realizada após quatro meses demonstrando áreas císticas decorrente de lesão vascular hipóxico-isquêmica. DISCUSSÃO Acalvaria primária é uma malformação congênita rara caracterizada pela ausência parcial ou completa dos ossos que formam a abóbada craniana, a musculatura associada e a dura-máter, com ossos faciais e da base do crânio normais. O defeito costuma estar recoberto por pele. O conteúdo intracraniano está completo, embora algumas anormalidades possam estar associadas(1-7). Sua etiologia e patogênese permanecem desconhecidas. Durante o desenvolvimento embrionário, após o fechamento do neuróporo anterior por volta da quarta semana, ocorre migração do mesênquima, porção membranosa do neurocrânio, abaixo do ectoderma. É dita secundária quando decorrente de síndrome de brida amniótica, defeitos do tubo neural ou do uso de inibidores da enzima conversora da angiotensina na gestação. Denomina-se hipocalvaria quando os ossos estão presentes, mas são hipoplásicos.(6) Pode estar associada a outras malformações como holoprosencefalia, hidrocefalia e micropoligiria, além de outras anomalias faciais, cardíacas, onfalocele, entre outras(1,6). Os poucos relatos sugerem uma predileção pelo sexo feminino com cariótipo normal(7), em concordância com a descrição deste caso. Observou-se presença de pele na região frontal da abóbada craniana e ausência parcial de couro cabeludo. A presença de pele e couro cabeludo, derivados do ectoderma, e a ausência de abóbada, musculatura associada e dura-máter, derivados da migração do mesênquima, caracterizam a acalvaria primária. Neste caso não se pode descartar alguma associação com um tipo extenso de aplasia cutis congenita. No mínimo, deve-se considerá-la no diagnóstico diferencial. Esta afecção é caracterizada pela ausência de formação completa da pele, o couro cabeludo é o local de ocorrência em 84% dos pacientes afetados por essa afecção, e o crânio é acometido em 15% a 30% das vezes. As lesões extensas e profundas são extremamente raras, com taxa de mortalidade elevada, e podem envolver o periósteo, o crânio e a dura-máter(8). REFERÊNCIAS 1. Harris CP, Townsend JJ, Carey JC. Acalvaria: a unique congenital anomaly. Am J Med Genet. 1993;46:694-9. 2. Raines C. Primary acalvaria. JDMS. 2006;22:407-10. 3. Bianca S, Ingegnosi C, Auditore S, et al. Prenatal and postnatal findings of acrania. Arch Gynecol Obstet. 2005;271:256-8. 4. Khadilkar VV, Khadilkar AV, Nimbalkar AA, et al. Acalvaria. Indian Pediatr. 2004;41:618-20. 5. Evans C, Marton T, Rutter S, et al. Cranial vault defects: the description of three cases that illustrate a spectrum of anomalies. Pediatr Dev Pathol. 2009;12:96-102. 6. Barr M, Cohen MM Jr. ACE inhibitor fetopathy and hypocalvaria. The kiney-skull connection. Teratology. 1991;44:485-95. 7. Raupp P, Nork M, Kappel I. Primary acalvaria in a term newborn. J Pediatr. 2002;141:593. 8. Yang JY, Yang WG. Large scalp and skull defect in aplasia cutis congenita. Br J Plast Surg. 2000;53:619-22. 1. Mestre, Coordenadora da Clínica de Imagem do Serviço de Obstetrícia e Ginecologia do Hospital Universitário da Universidade Federal do Maranhão (HU-UFMA), São Luís, MA, Brasil 2. Doutora, Chefe do Serviço de Obstetrícia e Ginecologia do Hospital Universitário da Universidade Federal do Maranhão (HU-UFMA), São Luís, MA, Brasil 3. Doutora, Chefe do Serviço de Neonatologia do Hospital Universitário da Universidade Federal do Maranhão (HU-UFMA), São Luís, MA, Brasil 4. Especialista, Médica Coordenadora da UTI-Neonatal do Serviço de Neonatologia do Hospital Universitário da Universidade Federal do Maranhão (HU-UFMA), São Luís, MA, Brasil 5. Especialista, Médica da UTI-Neonatal do Serviço de Neonatologia do Hospital Universitário da Universidade Federal do Maranhão (HU-UFMA), São Luís, MA, Brasil 6. Mestranda, Médica da UTI-Neonatal do Serviço de Neonatologia do Hospital Universitário da Universidade Federal do Maranhão (HU-UFMA), São Luís, MA, Brasil Trabalho realizado na Universidade Federal do Maranhão (UFMA), São Luís, MA, Brasil Endereço para correspondência: Dra. Lívia Teresa Moreira Rios Avenida do Vale, Ed. Costa Rica, ap. 801, Jardim Renascença 65075-820. São Luís, MA, Brasil E-mail: ltlrios@terra.com.br Recebido para publicação em 17/4/2010 Aceito, após revisão, em 24/5/2010 |
|
Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554