Radiologia Brasileira - Publicação Científica Oficial do Colégio Brasileiro de Radiologia
AMB - Associação Médica Brasileira CNA - Comissão Nacional de Acreditação
 Vol. 35 nº 5 - Set. / Out. of 2002
Vol. 35 nº 5 - Set. / Out. of 2002
|
RELATO DE CASO
|
|
|
|
|
Autho(rs): Celso Montenegro Turtelli |
|
|
Descritores: Mucopolissacaridoses, Síndrome de Maroteaux-Lamy, Gargolismo |
|
|
Resumo:
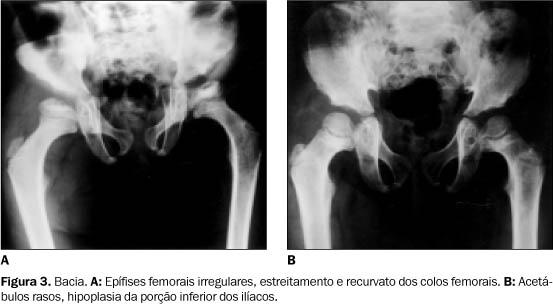
INTRODUÇÃO As mucopolissacárides são distúrbios hereditários de armazenamento lisossômico(1) em que há acúmulo de mucopolissacarídeos não-degradados em células e tecidos. Estas moléculas de alto peso molecular, também chamadas de glicosaminoglicanos, estão normalmente presentes na matriz celular(2). Por deficiências enzimáticas não são degradadas, acumulando-se em vários órgãos. Estes erros do metabolismo causam dez síndromes principais(1) e vários subtipos(2). As mais importantes, do ponto de vista clínico-radiológico, estão relatadas na Tabela 1. Todas as mucopolissacárides são caracterizadas por deterioração multissistêmica, crônica e progressiva, com alterações osteoarticulares, audiovisuais e cardiovasculares.
A síndrome de Mórquio e as mais raras, Scheie e Di Ferrante, são as únicas que não causam alterações do sistema nervoso central, como retardo mental ou retardo do desenvolvimento(3). Todas essas síndromes são do tipo autossômico recessivo, com exceção da mucopolissacáride II (Hunter), que é ligada ao cromossomo X(2). O termo gargolismo, dado a muitas dessas síndromes, é devido à fácies gargólica: "cabeça" grande, fácies grotesca, mão de garra, deformidade dos membros e pele espessa(2).
MATERIAL E MÉTODOS Relatamos, neste artigo, dois casos de mucopolissacárides tipo VI em pacientes irmãos, um deles do sexo masculino com sete anos de idade (caso A) e o outro do sexo feminino com cinco anos de idade (caso B). Ambos apresentavam fenótipo típico das mucopolissacárides: nanismo, cifose, deformidade das mãos, macrocrania e protrusão abdominal. Os pacientes foram examinados com radiografias do esqueleto axial, bacia, quadril, mãos e punhos. Em ambos também foi realizado estudo do encéfalo por ressonância magnética (RM). Os exames por RM foram realizados em aparelho de 0,5 T (Toshiba Flexart). As seqüências realizadas foram: T1, "FLAIR" e T2 (plano axial), T2 coronal e sagital. Não houve necessidade de injeção de contraste paramagnético (gadolínio).
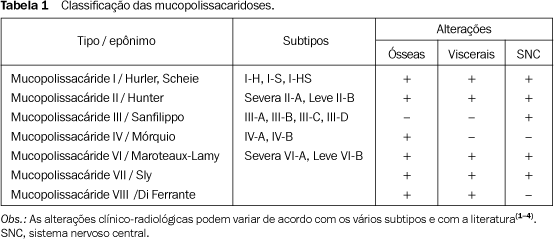
RESULTADOS As alterações radiográficas foram mais evidentes no caso A. Crânio ¾ No crânio notou-se aumento do diâmetro ântero-posterior, com abaulamento frontal, associado a sela turca em J (Figuras 1A e 1B).
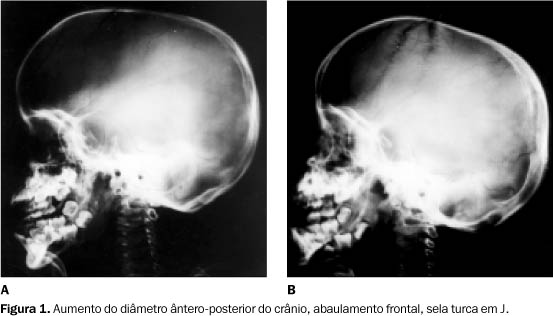
Coluna vertebral ¾ As principais alterações se relacionavam à configuração das vértebras e das costelas, evidenciando-se cifose toracolombar, corpos vertebrais hipoplásicos (ovalados) com protrusão óssea inferior, associados a pedículos alongados em perfil (Figuras 2A e 2B).
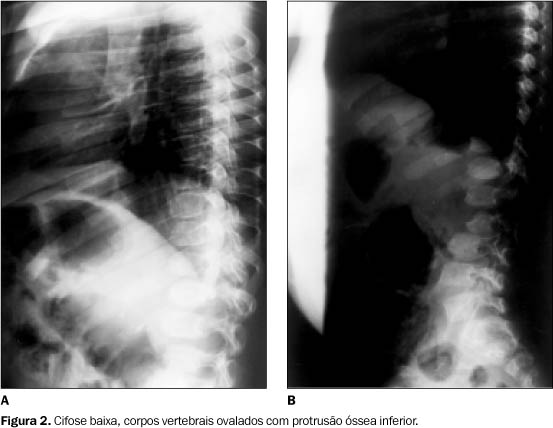
Bacia ¾ As diferenças radiográficas mais importantes entre os irmãos foram verificadas na região do quadril e bacia. No caso A evidenciaram-se densificação e irregularidade das epífises, com estreitamento e recurvato dos colos femorais (Figura 3A). Já no caso B, observou-se apenas hipoplasia dos ilíacos, inferiormente, com acetábulos rasos (Figura 3B).
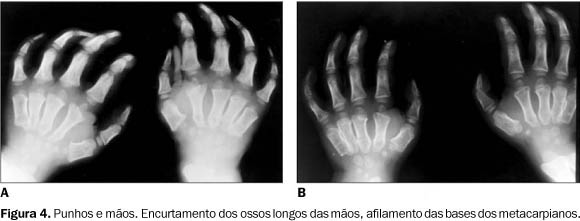
Punhos e mãos ¾ Nos punhos e mãos havia alargamento e encurtamento dos metacarpianos e falanges, acompanhados de afilamento das bases dos metacarpianos (Figuras 4A e 4B).
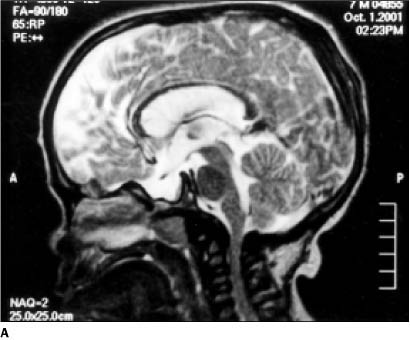
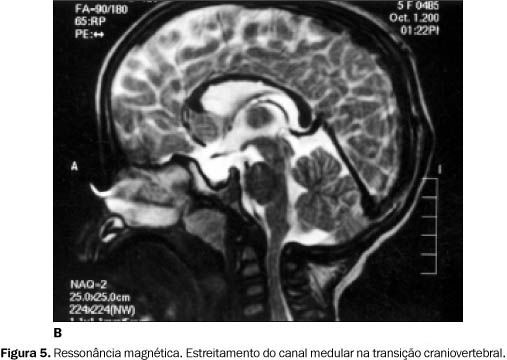
Os exames por RM evidenciaram, em ambos os casos, estreitamento do canal vertebral na transição craniovertebral (Figuras 5A e 5B).
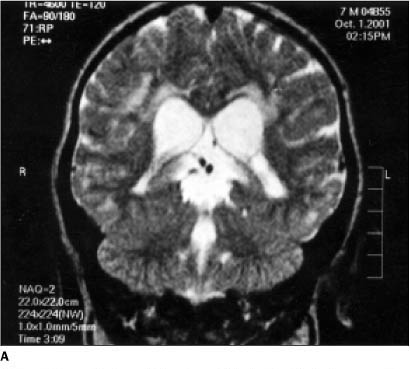
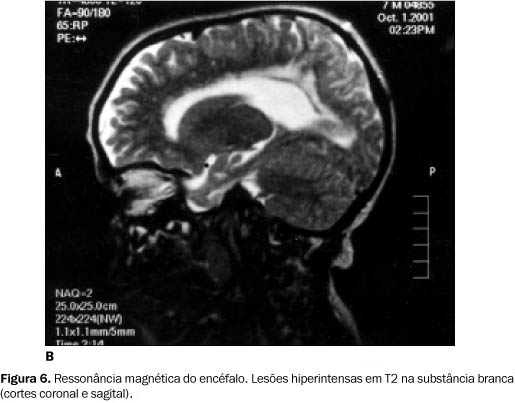
Apenas no caso A foram observadas alterações cerebrais tipo "bright-lesions". Estas alterações são devidas ao prolongamento do sinal em T1 e T2 na substância branca. As Figuras 6A e 6B evidenciam áreas hiperintensas em T2 na substância branca. O paciente do caso B não apresentou estas alterações, provavelmente pela menor idade.
DISCUSSÃO A mucopolissacáride tipo VI ou síndrome de Maroteaux-Lamy é caracterizada por alterações somáticas graves e poucas alterações mentais, o que a diferencia da síndrome de Hurler, na qual o fenótipo é semelhante, porém as alterações neurológicas são mais graves, com óbito antes dos dez anos de idade(2). As alterações clínico-radiológicas iniciam-se entre dois e quatro anos de idade, e as principais são: retardo de crescimento, cifose toracolombar, hepatoesplenomegalia e rigidez das articulações. A opacidade da córnea é precoce, decorrente do depósito de mucopolissacárides. As alterações na RM são variáveis. A principal é o estreitamento do canal vertebral na transição craniovertebral. Isto é devido a hiperostose e espessamento da dura-máter por acúmulo de mucopolissacárides. Em casos com alterações leves ou moderadas, pode haver espessamento da calota craniana e dilatação ventricular discreta. Com a evolução da doença, pode ocorrer alteração de sinal na substância branca periventricular(3). Isto se deve ao depósito de mucopolissacárides nos espaços perivasculares(4). O diagnóstico da síndrome de Maroteaux-Lamy é sugerido pelo exame de urina, que evidencia a presença de dermatansulfato e ausência do heparansulfato(5). Transplantes de medula óssea têm sido utilizados como método terapêutico nas mucopolissacárides. A RM é útil no controle pós-operatório destes pacientes, mostrando se há melhora na mielinização e maior contraste entre as substâncias branca e cinzenta(3).
CONCLUSÃO As mucopolissacárides em geral manifestam-se com deterioração multissistêmica progressiva e alterações radiológicas semelhantes. A gravidade das manifestações osteoarticulares, caracterizadas na radiografia, e neurológicas, na RM, auxiliam no diagnóstico e no prognóstico dessas síndromes. Orientam, quando necessário, o aconselhamento genético para a prevenção de novos casos(2). A RM também é importante no seguimento dos pacientes tratados com transplantes de medula(3).
REFERÊNCIAS 1.Lymm JB, John CC, Bamshad JM, Raymond WH, Motta PA. Genética médica. 2ª ed. Rio de Janeiro: Guanabara-Koogan, 2000:130¾2. [ ] 2.Diament A, Cypel S. Neurologia infantil. 3ª ed. São Paulo: Atheneu, 1996:474¾9. [ ] 3.Atlas WS. Magnetic resonance imaging of the brain and spine. 2nd ed. Philadelphia: Lippincott-Raven, 1996. [ ] 4.Murata R, Nakajima S, Tanaka A, et al. MR imaging of the brain in patients with mucopolysaccharidosis. AJNR 1989;10:1165¾70. [ ] 5.Menkes JH. Textbook of child neurology. 5th ed. Baltimore: Williams & Wilkins, 1995:chapt 1. [ ]
* Trabalho realizado no Serviço de Diagnóstico por Imagem do Hospital São Domingos, Uberaba, MG. |
|
Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554