Radiologia Brasileira - Publicação Científica Oficial do Colégio Brasileiro de Radiologia
AMB - Associação Médica Brasileira CNA - Comissão Nacional de Acreditação
 Vol. 35 nº 4 - Jul. / Ago. of 2002
Vol. 35 nº 4 - Jul. / Ago. of 2002
|
ARTIGO ORIGINAL
|
|
|
|
|
Autho(rs): Edson Marchiori, Simone Damato, Rosana Rodrigues, Paulo Marcos Valiante, Renato Gonçalves de Mendonça, Tizuko Miyagui, Miguel Abidon Aidê |
|
|
Descritores: Pneumonia intersticial linfocítica, Tomografia computadorizada de alta resolução, Anatomopatologia |
|
|
Resumo:
INTRODUÇÃO A pneumonia intersticial linfocítica (PIL) é uma doença benigna que faz parte de um espectro de doenças linfoproliferativas dos pulmões, caracterizada por infiltração difusa do interstício por uma mistura polimórfica de linfócitos, histiócitos e células plasmáticas(1-3). A PIL está freqüentemente associada a alterações sistêmicas, como a síndrome de Sjögren, a doença auto-imune da tireóide, a doença de Castleman e a síndrome da imunodeficiência adquirida (SIDA)(2,4), ou, mais raramente, ao lúpus eritematoso sistêmico e outras doenças de origem autoimune, incluindo a miastenia gravis, a anemia perniciosa, a hepatite crônica ativa, e outras(5). Neste trabalho foram avaliados os aspectos observados nas tomografias computadorizadas de alta resolução (TCAR) de dois pacientes com PIL comprovada, e feita a correlação com os aspectos anatomopatológicos.
CASUÍSTICA E MÉTODOS Foram revistas as tomografias computadorizadas de alta resolução de dois pacientes com diagnóstico anatomopatológico de pneumonia intersticial linfocítica, do sexo feminino e com 24 anos e 41 anos de idade, respectivamente. Ambos os exames foram feitos com a técnica de alta resolução, com cortes axiais de 2 mm de espessura, com 10 mm de incremento, dos ápices até as bases pulmonares. Estes exames foram correlacionados com os achados anatomopatológicos, a partir de material obtido de biópsias a céu aberto.
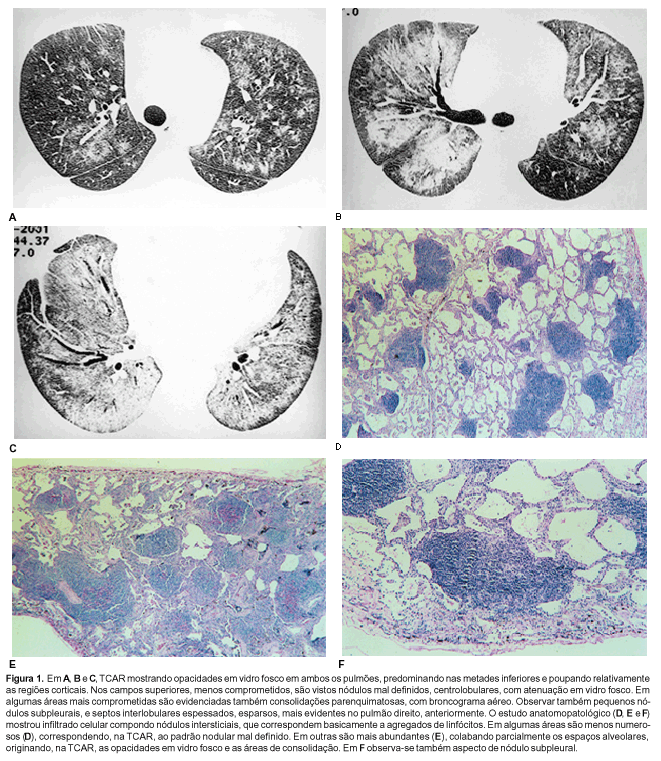
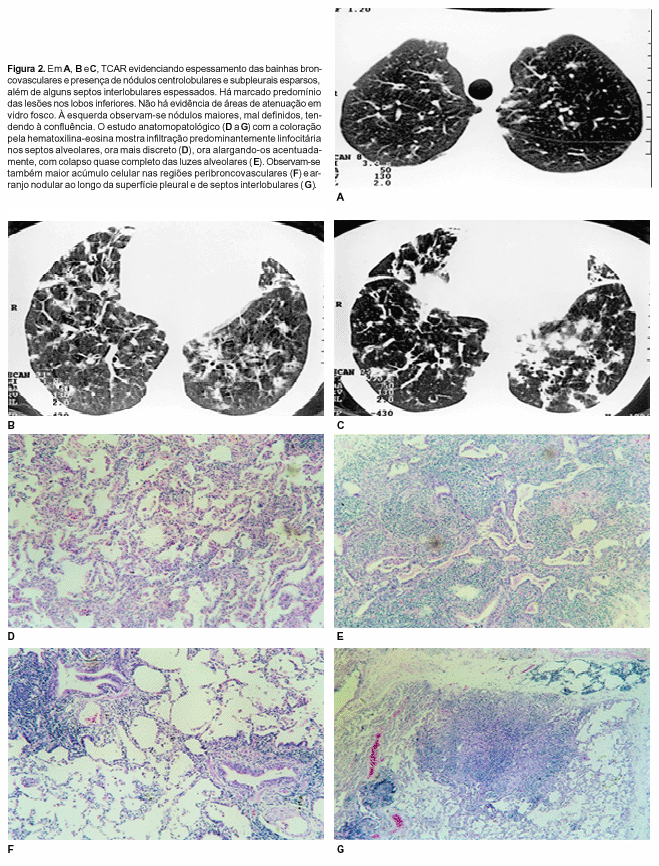
RESULTADOS Em um dos pacientes a TCAR (Figura 1) mostrou opacidades em vidro fosco em ambos os pulmões, predominando nas metades inferiores e poupando, relativamente, as regiões corticais. Nos campos superiores, menos comprometidos, foram vistos nódulos centrolobulares, mal definidos, com atenuação em vidro fosco. Em algumas áreas mais comprometidas foram evidenciadas consolidações parenquimatosas,com broncograma aéreo. Foram observados, também, pequenos nódulos subpleurais, e septos interlobulares espessados, esparsos, mais evidentes no pulmão direito, anteriormente. No outro paciente (Figura 2) o aspecto predominante foi de espessamento das bainhas peribroncovasculares e de septos interlobulares, predominando nos campos inferiores. Nódulos centrolobulares esparsos e nódulos subpleurais também foram identificados Na base do pulmão esquerdo havia nódulos maiores, mal definidos, tendendo à confluência. Cistos não foram identificados em nenhum dos dois pacientes. O estudo histopatológico revelou, nos dois casos, comprometimento principalmente dos septos alveolares, que se encontravam espessados por denso infiltrado predominantemente linfocitário, embora, em menor quantidade, houvesse também histiócitos, plasmócitos e eosinófilos de permeio. Em algumas áreas o infiltrado era mais discreto e em outras os septos alveolares estavam espessados a tal ponto que praticamente colabavam as luzes alveolares. Em um dos casos, em alguns campos já se observavam também algum grau de fibrose nos septos alveolares e hiperplasia de pneumócitos. Em outros campos notavam-se fibrose mais amadurecida e infiltração mononuclear mais intensa apagando o desenho alveolar e restringindo os espaços aéreos a pequenas fendas, internamente revestidas por pneumócitos jovens. Em alguns pontos até mesmo estas estruturas desapareciam. Os campos vizinhos apresentavam dilatação alveolar irregular. O quadro demonstra tendência à destruição parenquimatosa, resultando em faveolamento. No outro caso o infiltrado linfocitário apresentava, de permeio, folículos linfóides hiperplasiados, com centros germinativos. Em algumas áreas o infiltrado predominava no interstício peribroncovascular. As células inflamatórias mostraram reação positiva para as imunoglobulinas de cadeia leve kappa e lambda, linfócitos T e histiócitos.
DISCUSSÃO A PIL é mais comum em mulheres na quinta década de vida, embora a idade possa variar de meses até 80 anos(5). Clinicamente, os pacientes se apresentam com tosse e dispnéia progressiva(6), podendo haver também febre, perda de peso, artralgias e dor pleurítica(5). Alguns pacientes cursam com quadro de infecções de repetição, tanto de vias aéreas superiores como de vias inferiores. O achado laboratorial mais constante é a disgamaglobulinemia, em geral uma gamopatia policlonal difusa, embora gamopatia monoclonal já tenha sido descrita(7). Nos nossos casos hipogamaglobulinemia policlonal foi observada. A história natural da PIL é muito variada. Em alguns pacientes a doença involui após tratamento com corticosteróides, en quanto em outros ela fica estável ou progride, apesar do tratamento, com deterioração progressiva da função pulmonar e evolução para pulmão em estágio terminal, com fibrose e faveolamento(3,5,6). Desenvolvimento de linfomas tem sido relatado em alguns casos, mas acredita-se que estes pacientes tinham linfoma maligno desde o início(3). A maior parte dos casos é em adultos, mas também pode ocorrer em crianças. Crianças com PIL freqüentemente estão infectadas pelo vírus da imunodeficiência humana(6,8). A PIL é vista em 30% a 40% das crianças com SIDA. Ela em geral tem curso crônico, com prognóstico relativamente bom(8). Johkoh et al.(2) fizeram estudo retrospectivo das TCAR de 22 pacientes com PIL confirmada histopatologicamente. Neste estudo, atenuação em vidro fosco e nódulos centrolobulares foram observados em todos os pacientes. Estas lesões eram bilaterais, envolvendo todas as regiões dos pulmões. Embora o processo seja difuso, a PIL tem tendência a ser mais acentuada no interstício perilinfático, isto é, ao longo das bainhas broncovasculares, septos interlobulares e pleura(2). Desta forma, espessamento peribroncovascular, de septos interlobulares e pequenos nódulos subpleurais foram vistos na maioria dos pacientes. Cistos pulmonares foram encontrados em 68% dos casos. Ichikawa et al.(9) sugeriram que o mecanismo para a formação dos cistos fosse a presença de infiltrados linfocitários nodulares, peribroncovasculares, com obstrução parcial da via aérea. Linfonodos aumentados são freqüentes, mas é questionável o quanto se devem às doenças associadas (doença de Castleman, síndrome de Sjögren, SIDA, etc.)(2). Em crianças o aspecto tomográfico mais comum é a infiltração difusa, com padrão em vidro fosco. Consolidação do espaço aéreo e eventual desenvolvimento de bronquiectasias também têm sido observados, além de formações císticas, por vezes associadas a pneumotórax. Freqüentemente estas alterações se acompanham de linfonodomegalias mediastinais e/ou hilares, podendo atingir grandes dimensões(8). Microscopicamente, o pulmão mostra infiltrado celular predominantemente intersticial, que compromete difusamente o parênquima distal. O infiltrado é composto por uma mistura de pequenos linfócitos maduros, células plasmáticas e histiócitos, que alargam os septos alveolares e envolvem pequenas vias aéreas e vasos(6). Nódulos linfóides intersticiais, freqüentemente contendo centros germinativos, são comuns, com graus variados de fibrose intersticial e coleções mal definidas de histiócitos epitelióides lembrando granulomas(5). Por vezes o infiltrado celular derrama-se para os espaços alveolares, e pequeno número de linfócitos e histiócitos pode se acumular nestas áreas(6). Nos bronquíolos terminais e respiratórios os agregados linfóides comprimem as vias aéreas, produzindo graus variados de estenose bronquiolar. Em cerca da metade dos casos obstrução bronquiolar resulta de massas luminais de tecido conjuntivo mixóide jovem, misturado com células inflamatórias, reproduzindo um padrão de bronquiolite obliterante. Como resultado da obstrução, grande número de células (principalmente macrófagos e pneumócitos) ficam coletadas nos sacos alveolares(1). Embora a PIL possa mostrar distribuição peribrônquica e perivascular, não há infiltração vascular, destruição brônquica ou lesão linfoepitelial. A lesão também se caracteriza por ausência de enchimento alveolar significativo(10). O diagnóstico diferencial deve ser feito com pneumonite por hipersensibilidade, com pneumocistose no paciente com SIDA, e com outras doenças linfoproliferativas(2). Recentes avanços nas técnicas imuno-histoquímicas têm sugerido que vários dos casos inicialmente diagnosticados com PIL correspondiam a proliferações monoclonais, devendo ser considerados linfomas não-Hodgkin de baixo grau(10). A diferenciação com os linfomas pode ser complicada, uma vez que eventualmente as duas doenças têm características clínicas, radiológicas e até anatomopatológicas bastante semelhantes(4). Técnicas de imunofenotipagem e imuno-histoquímica do tecido pulmonar biopsiado são muito úteis para esta diferenciação. Também na TCAR o diagnóstico diferencial pode ser bastante difícil. Os cistos são característicos da PIL, enquanto as consolidações, grandes nódulos e derrames pleurais são característicos dos linfomas malignos. Pequenos nódulos, opacidades em vidro fosco e linfonodomegalias hilares e/ou mediastinais são encontrados nas duas doenças, sem diferenças estatisticamente significativas(4,11). Estudo realizado por Johkoh et al.(3), avaliando a evolução das lesões em 14 pacientes, em tempos que variaram de 4 a 82 anos (média de 13 meses), constatou que nove melhoraram, um ficou inalterado e quatro pioraram. Com exceção dos cistos, as alterações parenquimatosas foram todas reversíveis. Novos cistos foram encontrados, em geral se desenvolvendo em áreas onde previamente existiam nódulos centrolobulares. Faveolamento foi observado em quatro pacientes, três deles em áreas de consolidação prévias. Em resumo, a maior parte das lesões melhora com o tratamento. No entanto, consolidações do espaço aéreo podem evoluir para faveolamento, e nódulos centrolobulares podem preceder a formação de cistos(3).
REFERÊNCIAS 1.Oldham SAA, Castillo M, Jacobson FL, Mones JM, Saldana MJ. HIV-associated lymphocytic interstitial pneumonia: radiologic manifestations and pathologic correlation. Radiology 1989;170(1 Pt 1):83-7. [ ] 2.Johkoh T, Muller NL, Pickford HA, et al. Lymphocytic interstitial pneumonia: thin-section CT findings in 22 patients. Radiology 1999;212:567-72. [ ] 3.Johkoh T, Ichikado K, Akira M, et al. Lymphocytic interstitial pneumonia: follow-up CT findings in 14 patients. J Thorac Imaging 2000;15:162-7. [ ] 4.Honda O, Johkoh T, Ichikado K, et al. Differential diagnosis of lymphocytic interstitial pneumonia and malignant lymphoma on high-resolution CT. AJR 1999;173:71-4. [ ] 5.Vath RR, Alexander CB, Fulmer JD. The lymphocytic infiltrative lung diseases. Clin Chest Med 1982;3:619-34. [ ] 6.Katzenstein ALA, Askin FB, Katzenstein AL, Day L, Livolsi VA. Katzenstein and Askin's Surgical pathology of non-neoplastic lung disease. 3rd ed. Philadelphia: WB Saunders, 1997. [ ] 7.Kradin RL, Mark EJ. Benign lymphoid disorders of the lung, with a theory regarding their development. Hum Pathol 1983;14:857-67. [ ] 8.Marks MJ, Haney PJ, McDermott MP, White CS, Vennos AD. Thoracic disease in children with AIDS. Radiographics 1996;16:1349-62. [ ] 9.Ichikawa Y, Kinoshita M, Koga T, Oizumi K, Fujimoto K, Hayabuchi N. Lung cyst formation in lymphocytic interstitial pneumonia: CT features. J Comput Assist Tomogr 1994;18:745-8. [ ] 10.McGuinness G, Scholes JV, Jagirdar JS, et al. Unusual lymphoproliferative disorders in nine adults with HIV or AIDS: CT and pathologic findings. Radiology 1995;197:59-65. [ ] 11.Webb WR, Muller NL, Naidich DP. High-resolution CT of the lung. 3rd ed. Philadelphia: Lippincott Williams & Wilkins, 2001:138-45. [ ]
* Trabalho realizado nos Serviços de Radiodiagnóstico e de Anatomia Patológica do Hospital Universitário Clementino Fraga Filho (HUCFF) da Universidade Federal do Rio de Janeiro (UFRJ), Rio de Janeiro, RJ, e nos Departamentos de Radiologia, de Patologia e de Medicina Clínica da Universidade Federal Fluminense (UFF), Niterói, RJ. |
|
{kind=link}
{kind=link}
Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554