Radiologia Brasileira - Publicação Científica Oficial do Colégio Brasileiro de Radiologia
AMB - Associação Médica Brasileira CNA - Comissão Nacional de Acreditação
 Vol. 36 nº 4 - Jul. / Ago. of 2003
Vol. 36 nº 4 - Jul. / Ago. of 2003
|
RELATO DE CASO
|
|
|
|
|
Autho(rs): Sizenildo da Silva Figueirêdo, Renato Santos Argollo, Marlos Augusto Bittencourt Costa, Flávia Aparecida de Souza Ribeiro, Bruno Barcelos da Nóbrega, Sebastião Alves Pinto, Leonardo Valadares Barbosa Lôbo, Marise Amaral Moreira Rebolças, Kim-Ir-Sen Santos Teixeira |
|
|
Descritores: Carcinoma de células renais, Carcinoma renal sarcomatóide, Metaplasia tumoral, Tomografia computadorizada, Ultra-sonografia |
|
|
Resumo: IIIMédico Residente do Serviço de Diagnóstico por Imagem do HC-FMUFG
INTRODUÇÃO O rim, resultado de um complexo desenvolvimento embriológico, apresenta uma grande variedade de tumores, alguns dos quais têm equivalentes em outros órgãos, porém alguns sendo exclusivos do sítio renal(1). O carcinoma de células renais (CCR) é o tumor maligno mais comum do rim(2). Compreende 3% dos cânceres em adultos(3) e corresponde a um adenocarcinoma com variações histológicas dos tipos células claras ou convencional (70%), papilar (15% a 20%), células granulares (7%), células cromófobas (6% a 11%), células do ducto coletor (menos que 1%) e o subtipo sarcomatóide(4). A variante sarcomatóide do CCR ocorre em 1% a 13% dos casos(5?7), segundo algumas séries. Inicialmente acreditava-se que um crescimento simultâneo de células epiteliais e estromais originavam esses tumores sarcomatóides incomuns, sendo erroneamente classificados como carcinossarcomas(5). A observação histológica da presença de células renais malignas sofrendo graves transformações metaplásicas para um padrão sarcoma-símile resultou na criação da denominação "carcinoma renal sarcomatóide" (CRS)(6). O CRS consiste em um CCR associado mais comumente a células fusiformes pleomórficas similares às do fibrossarcoma, cujos focos de transição entre células carcinomatosas e células sarcomatóides podem ser comumente demonstrados(7). Além do CCR convencional, a metaplasia sarcomatóide também tem sido descrita no carcinoma renal papilar, no carcinoma dos ductos coletores e em séries de carcinoma renal cromófobo(8?10). O CRS possui atividade proliferativa muito intensa, determinando péssimo prognóstico, pois seu diagnóstico comumente é realizado apenas em estágios avançados(11), em função da pobreza de manifestações clínicas iniciais. Comparação entre CRS e CCR convencional revela similaridade quanto a faixa etária, sexo e distribuição topográfica no rim, porém com diferenças no comportamento biológico e repercussões clínicas(5) Os vários métodos de diagnóstico por imagem podem fornecer achados importantes para diferenciar o CRS de tumores sarcomatosos verdadeiros(5,12). A utilização desses métodos auxilia no estadiamento pré-operatório, objetivando delinear as margens tumorais, infiltração de estruturas vasculares, linfonodais, bem como na pesquisa de implantes metastáticos. Relatamos, neste trabalho, um caso de CRS, enfatizando os aspectos revelados pelos métodos de imagem e pela anatomia patológica inerentes a esta neoplasia, bem como destacando a necessidade de um correto diagnóstico para a determinação de abordagem terapêutica e de análise prognóstica mais precisas.
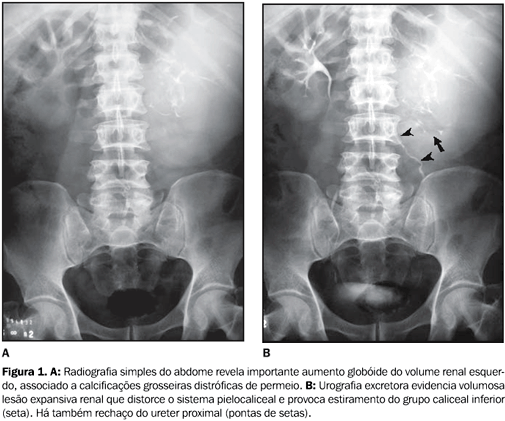
RELATO DO CASO Paciente do sexo masculino, 54 anos de idade, foi atendido no Serviço de Urologia do Hospital das Clínicas da Universidade Federal de Goiás, Goiânia, GO, com quadro clínico iniciado três meses antes da consulta, consistindo em hematúria macroscópica, dor lombar à esquerda, fraqueza e perda ponderal de aproximadamente 10 kg. Ao exame físico evidenciou-se massa palpável no flanco esquerdo, de aproximadamente 20 × 15 cm de diâmetro, de consistência endurecida e aderida aos planos profundos. A avaliação radiológica por urografia excretora demonstrou: acentuado aumento volumétrico do rim esquerdo com calcificações distróficas e irregulares no seu interior; retardo na concentração e eliminação do meio de contraste iodado; presença de cálices renais identificáveis apenas na porção inferior do órgão (os quais estavam deslocados, alongados e distorcidos) e ureter ipsilateral pérvio e anatômico, porém rechaçado (Figura 1). O rim contralateral não mostrou anormalidades.
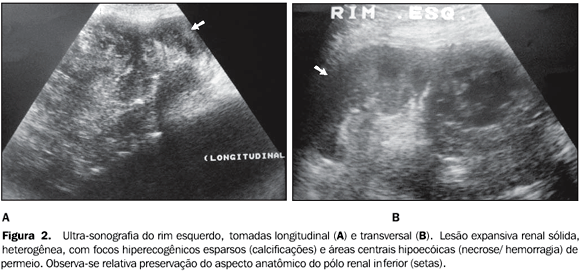
A ultra-sonografia (US) delineou com maior precisão os achados presentes à urografia excretora, mostrando o rim esquerdo bastante aumentado de volume, com distorção de sua arquitetura normal, à custa de volumosa lesão expansiva eco-complexa, de contornos lobulados, com áreas anecóicas e focos de calcificação grosseiros de permeio. A quase totalidade do rim estava acometida, sendo observado parênquima preservado apenas em seu pólo inferior (Figura 2).
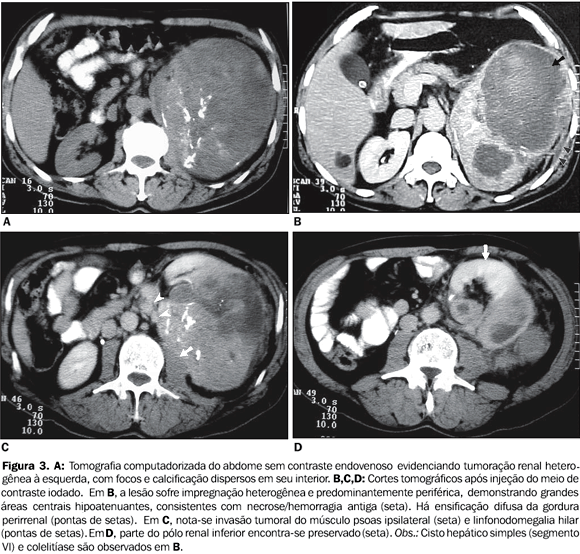
A tomografia computadorizada (TC), além de ratificar os achados dos exames anteriores, forneceu dados importantes a respeito do grau de vascularização da lesão, mostrando impregnação heterogênea do meio de contraste, com realce periférico das áreas necróticas. Permitiu, também, avaliar a extensão tumoral e infiltração das estruturas adjacentes, revelando invasão da gordura perirrenal e do músculo psoas ipsilaterais. A veia renal esquerda aparentemente estava preservada e linfonodomegalia hilar ipsilateral foi evidenciada (Figura 3). Não havia focos metastáticos nos demais órgãos abdominais, porém múltiplos implantes secundários nodulares foram vistos nas bases pulmonares e verificados também à radiografia de tórax, configurando estádio IV pré-cirúrgico.
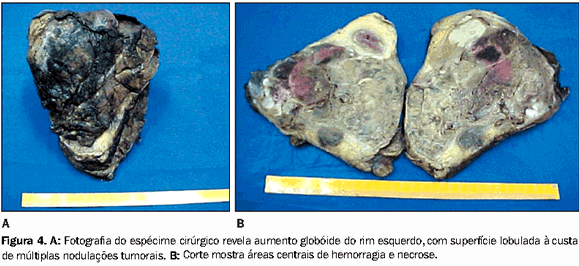
Nefrectomia total esquerda foi realizada, retirando-se, conjuntamente, a lesão de 2.500 g, medindo 28,0 × 26,0 × 22,0 cm nos maiores diâmetros, de limites capsulares evidentes, coloração branco-amarelada, com áreas de hemorragia, de necrose e calcificações irregulares dispersas. Havia invasão da gordura perirrenal e comprometimento de linfonodos peri-hilares. A porção proximal da veia renal estava parcialmente comprometida (Figura 4).
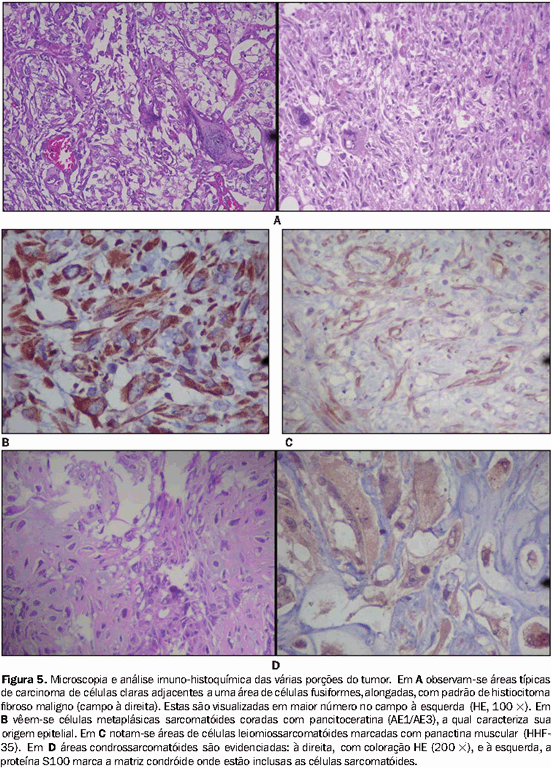
O diagnóstico histopatológico foi de CRS composto por áreas com padrão de carcinoma de células claras e outras com aspecto de fibro-histiocitoma maligno, de leiomiossarcoma e de condrossarcoma. A análise imuno-histoquímica das áreas sarcomatóides com múltiplos marcadores confirmou a origem epitelial em função da positividade destas para a citoceratina (AE1/AE3) e caracterizou os vários padrões sarcoma-símile (Figura 5).
Atualmente o paciente encontra-se em acompanhamento ambulatorial e sob quimioterapia sistêmica complementar.
DISCUSSÃO A denominação "carcinoma renal sarcomatóide" foi inicialmente proposto por Farrow et al.(6), em 1968, após a análise de 37 casos de malignidade renal caracterizados por um padrão misto de células neoplásicas. O entendimento da patogênese do CRS foi obtido pela observação de áreas tumorais com morfologia transicional entre carcinomas e sarcomas. Esses focos transicionais forneceram a evidência de serem as áreas sarcomatosas adjacentes ao CCR resultado da transformação metaplásica de células epiteliais malignas e, portanto, a denominação escolhida foi considerada mais apropriada(6,9). O CRS é neoplasia relativamente rara, cuja natureza não havia sido destacada nas primeiras classificações das malignidades renais, sendo inicial e erroneamente considerado sarcoma verdadeiro, derivado de fibroblastos e células musculares lisas do interstício renal ou da cápsula(9). As características macroscópicas do CRS não são diagnósticas desta neoplasia, sendo o CRS praticamente indistinguível do CCR convencional(8). Esses tumores são predominantemente parenquimatosos, não encapsulados, localmente invasivos(5,6), comumente de tamanho grande e com diâmetro médio de 10 cm(5,6,13,14). Essas lesões costumam mostrar aspecto heterogêneo, com áreas amareladas, friáveis, multinodulares e ocasionalmente hemorrágicas de CCR convencional adjacentes a outras regiões com aspecto mais sólido e fibroso, típico dos componentes sarcomatóides(5). A proporção de componentes sarcomatosos e carcinomatosos pode variar em diferentes tumores e ambas as regiões podem ser bem demarcadas ou indistintas entre si(5,15). O aspecto microscópico típico dos CRS inclui tumor misto com padrão dimórfico contendo vários focos de elementos sarcoma-símile de alto grau de anaplasia associados aos clássicos componentes epiteliais malignos interpostos ou adjacentes(5). A histologia das porções carcinomatosas do CRS apresenta padrão epitelióide que pode ser comum a qualquer tipo de CCR, incluindo tumores de células claras, cromófilos ou papilares, cromófobos, carcinoma dos ductos coletores ou subtipos sem classificação definida(5,8,13,14). Os componentes carcinomatosos de células claras ou cromófilos são os mais comumente encontrados(5,13,14). A histologia do componente sarcomatóide pode variar, porém a presença de células fusiformes pleomórficas e/ou células gigantes é vista na maioria dos tumores(5,15). Grande parte dos CRS mostra mitoses abundantes e aberrantes(5,13), alto grau de pleomorfismo celular(5,9,13) e extensas áreas de necrose confinadas geralmente à região sarcomatosa(5,13). Variantes sarcomatóides têm sido descritas. Os tipos mais comuns assemelham-se ao fibrossarcoma ou ao histiocitoma fibroso maligno(5,6,13). Menos comumente, o CRS pode assemelhar-se a rabdomiossarcoma(5,6,9), hemangiopericitoma(5,13,15), sarcoma osteogênico(5,6,16) ou conter focos de tecido mixóide com células gigantes multinucleadas osteoclasto-símile em um fundo de elementos sarcomatosos(5,9,13). Raramente os CRS contêm áreas de osteóide ou trabéculas ósseas mineralizadas em associação a camadas mais típicas de células fusiformes malignas e a focos de cartilagem maligna. Tais tumores têm sido denominados CRS osteogênicos, e na grande parte dos menos de 20 casos reportados até o ano de 2000, áreas de típico CCR convencional foram encontradas adjacentes às células sarcomatóides(6,9,17,18). A imuno-histoquímica também fornece evidência da patogênese dessas lesões, revelando tumores que expressam variáveis marcadores epiteliais. Em muitas séries a anticitoceratina AE1/AE3 foi encontrada em quase todos os casos(3,5), sendo o mais sensível marcador da natureza epitelial metaplásica dessas lesões(3). Antígenos de membrana epitelial estão presentes nas células fusiformes malignas em cerca de 50%(3) dos casos e a expressão da vimentina é geralmente mais prevalente nos elementos sarcomatóides que nos carcinomatosos(3). Os principais aspectos anatomopatológicos foram coincidentes ao nosso caso, porém a presença de três subtipos sarcomatóides (histiocitoma fibroso maligno, leiomiossarcoma e condrossarcoma) em uma única lesão torna este caso ainda mais raro em comparação à maioria dos já publicados, principalmente em função da presença das áreas condrossarcomatóides. Pacientes com CRS geralmente apresentam-se com sinais e sintomas típicos de CCR convencional. Os mais comuns são idênticos à tríade clássica do CCR: dor abdominal ou no flanco, tumoração palpável e hematúria(5,6,13). Não têm sido reportados casos de CRS bilaterais(5). A maioria dos CRS é encontrada em pacientes idosos e masculinos, semelhantemente ao CCR convencional(9). A média de idade varia de 56 a 61 anos(9). O CRS é o mais agressivo subtipo de CCR, apresentando péssimo prognóstico na atualidade(7). Este está diretamente relacionado a critérios anatomopatológicos detalhadamente estudados. Poucos CRS estão confinados à cápsula renal por ocasião do diagnóstico(5) e vários estudos têm mostrado que o estádio patológico é o fator de prognóstico isolado mais importante(5,14,15), com a sobrevida média para o estádio I de 50 meses e de apenas 4?5 meses para os estádios III e IV. Outros fatores associados com pior prognóstico incluem o envolvimento linfonodal e metástases a distância(5,12). Outros aspectos histológicos, tais como padrão de crescimento sarcomatóide, nível de celularidade, grau de fibrose e a quantidade dos elementos sarcomatóides, aparentemente não manifestam influência prognóstica(5). Os CRS são radiologicamente indistinguíveis de outros carcinomas renais, mas podem ser suspeitados na vigência de lesões altamente agressivas(19). À radiografia simples, os grandes tumores infiltrantes podem causar aumento de todo o contorno renal. Podem deslocar o rim completamente e até mesmo cruzar a linha mediana(19). Focos de calcificação com aspecto curvilíneo, puntiforme ou flocular(19) também podem ser evidenciados. À urografia excretora, esses grandes tumores costumam causar distorção do contorno renal normal e deslocamento do sistema coletor(19). Ocasionalmente, os carcinomas renais invadem os cálices ou a pelve renal, causando falhas de enchimento liso ou irregular, podendo também obstruir o sistema coletor(19). A ausência de eliminação do meio de contraste pelo rim contendo um carcinoma usualmente indica oclusão da veia renal por comprometimento tumoral, mas pode ser decorrente de uma extensa infiltração renal ou obstrução na junção pieloureteral pelo tumor(19). À US, os maiores CCR costumam apresentar contornos lobulados e textura heterogênea. Focos hiperecogênicos produtores de sombra acústica posterior correspondem a calcificações intratumorais(20). A invasão do sistema coletor não é rotineiramente detectada pela US e o envolvimento de linfonodos hilares é normalmente identificado(20). A extensão extracapsular do tumor e a invasão da gordura perirrenal são identificáveis com maior facilidade pela TC(20). Estes últimos aspectos não foram fielmente obtidos na análise ecográfica de nosso caso, sendo imprescindível o estudo da lesão por meio de TC. A TC é o método de escolha no diagnóstico e estadiamento do tumor renal. O aspecto do tumor pela TC e sua repercussão sobre o rim e estruturas adjacentes dependerão de seu tamanho, localização e composição(20). Os tumores sarcomatóides tendem a ser heterogêneos. As áreas císticas representam necrose ou hemorragia antiga e as sólidas, regiões mais celulares do tumor(20). Focos hiperdensos podem corresponder a hemorragias recentes e calcificações são identificadas e delineadas com precisão pela TC(20), estando presentes em cerca de 10% dos casos(12). Na fase contrastada, o CRS geralmente mostra-se com um realce heterogêneo ou predominantemente periférico(4). O meio de contraste iodado torna mais evidente as áreas císticas, de tecido necrótico ou de hemorragias antigas, pois estas não se impregnam(20). A evidenciação de trombos tumorais na veia cava inferior e veias renais também pode ser feita pela TC. O trombo tumoral é identificado como uma falha de enchimento na luz do vaso e habitualmente o calibre do vaso pode estar aumentado(20). Quando o trombo adquire vascularização própria por meio de capilares arteriais malformados, a TC é capaz de demonstrar a impregnação do trombo na fase pós-contraste imediato(20). Em nosso caso não conseguimos identificar trombos tumorais na veia renal ipsilateral, a qual apresentava calibre e opacificação usuais, apesar de conter pequenos focos de infiltração neoplásica em sua porção proximal à análise anatomopatológica. Quanto à disseminação linfática do tumor renal, linfonodos com mais de 1 cm de diâmetro são considerados suspeitos e quando maiores de 2 cm de diâmetro quase sempre são metastáticos(20). Esta disseminação se faz inicialmente para os linfonodos do hilo renal e depois para outros do retroperitônio, ao redor da aorta e da veia cava inferior. A infiltração perirrenal e de estruturas e órgãos vizinhos também ficará mais evidente após injeção do contraste, particularmente se for possível realizar-se TC dinâmica ou angio-TC(20). A ressonância magnética (RM) ratifica os achados tomográficos, mostrando massa irregular com margens mal definidas e realce heterogêneo em imagens pós-gadolínio imediatas que diminui em fases pós-contraste mais tardias(2). São tumores freqüentemente hipervasculares que rapidamente apresentam um "washout" do contraste, enquanto o córtex normal evidencia hipersinal devido à retenção de contraste nos túbulos renais(2). Áreas de hemorragia comumente apresentam hipersinal nas seqüências ponderadas em T1(20). Calcificações tumorais não são satisfatoriamente detectadas pela RM(20), e os linfonodos acometidos possuem sinal de média intensidade em T1 e alta intensidade em T2, não sendo possível, por este método, distinguir linfonodomegalia por infiltração tumoral de linfonodomegalia por hiperplasia reativa. A RM também tem sido útil na avaliação de trombos tumorais ou extensão do tumor aos órgãos adjacentes. Trombos são bem evidenciados em imagens spin-eco devido ao elevado contraste entre o trombo e a ausência de sinal do sangue nestas seqüências(2). A boa demonstração de trombose vascular ou infiltração tumoral de órgãos ou estruturas vizinhas pela RM revela ser este método de acuidade semelhante à TC(20). A RM está indicada quando houver antecedente de alergia ao meio de contraste iodado usado em TC(20). Os sítios metastáticos mais comuns são os pulmões e ossos (particularmente pelve e fêmur)(5,11,13,14), porém qualquer órgão pode ser afetado. Normalmente, implantes em órgãos distantes estão presentes devido ao estágio avançado quando os tumores são descobertos e ao comportamento muito agressivo do CRS. Embora os métodos de imagem sejam úteis para tentar diferenciar os sarcomas verdadeiros do CRS, não podem ser utilizados para diferenciar o CRS do CCR convencional, sendo estes praticamente indistinguíveis(5,12). O CRS caracteristicamente surge do parênquima renal, é consistentemente hipervascularizado em áreas não necróticas, possui abundante neovascularização e freqüentemente estende-se às veias renal e cava inferior(5). Também pode ter extensão direta às regiões para-aórticas e estar comumente associado a linfonodomegalia retroperitoneal(5,12). Os sarcomas verdadeiros, tumores extremamentes raros(3), opostamente, tendem a se originar da cápsula renal ou do seio renal, permanecem confinados à cápsula, sem envolvimento das veias renal ou cava inferior, geralmente são hipovasculares e a presença de tecido adiposo sugere tratar-se de um lipossarcoma, nesse contexto(5,12). Em nosso caso foram observadas extensão aos linfonodos hilares e invasão do músculo psoas, refletindo a agressividade da lesão e sugerindo um CRS também como hipótese diagnóstica. A conjunção dos vários métodos de diagnóstico por imagem torna mais precisa a abordagem cirúrgica das lesões malignas renais ao auxiliar no seu estadiamento, bem como sugerir as hipóteses diagnósticas mais prováveis frente às suas características. Apesar da relatada raridade do CRS, a sua confirmação pelo estudo histológico prediz um mau prognóstico e sugere ao médico urologista as reais expectativas de sobrevida do paciente, mesmo diante das avançadas abordagens terapêuticas existentes atualmente.
REFERÊNCIAS 1. Farrow GM, Harrison EG Jr, Utz DC, ReMine WH. Sarcomas and sarcomatoid and mixed malignant tumors of the kidney in adults. Part I. Cancer 1968; 22:545?50. [ ] 2. Semelka RC, Kelekis NL. Kidneys. In: Semelka RC, Ascher SM, Reinhold C, eds. MRI of the abdomen and pelvis ? a text atlas. New York: Wiley-Liss, 1997:379?470. [ ] 3. DeLong W, Grignon DJ, Eberwein P, Shum DT, Wyatt JK. Sarcomatoid renal cell carcinoma. An immunohistochemical study of 18 cases. Arch Pathol Lab Med 1993;117:636?40. [ ] 4. Kim JK, Kim TK, Ahn HJ, Kim CS, Kim KR, Cho KS. Differentiation of subtypes of renal cell carcinoma on helical CT scans. AJR 2002;178:1499?506. [ ] 5. Chao D, Zisman A, Freeland SJ, Pantuck AJ, Said JW, Belldegrun AS. Sarcomatoid renal cell carcinoma: basic biology, clinical behavior and response to therapy. Urol Oncol 2001;6:231?8. [ ] 6. Farrow GM, Harrison EG Jr, Utz DC. Sarcomas and sarcomatoid and mixed malignant tumors of the kidney in adults. Part III. Cancer 1968;22:556?63. [ ] 7. Tomera KM, Farrow GM, Lieber MM. Sarcomatoid renal carcinoma. J Urol 1983;130:657?9. [ ] 8. Akhtar M, Tulbah A, Kardar AH, Ali MA. Sarcomatoid renal cell carcinoma: the chromophobe connection. Am J Surg Pathol 1997;21:1188?95. [ ] 9. Delahunt B. Sarcomatoid renal carcinoma: the final common dedifferentation pathway of renal epithelial malignancies. Pathology 1999;31:185?90. [ ] 10. Aizawa S, Chigusa M, Ohno Y, Suzuki M. Cromophobe cell renal carcinoma with sarcomatoid component. A report of two cases. J Urol Pathol 1997;6: 51?9. [ ] 11. Magallon GH, Arnaut AS. Carcinoma renal sarcomatoide: caso clínico y revisión de la literatura. Med Int Mex 2000;16:43?5. [ ] 12. Shirkhoda A, Lewis E. Renal sarcoma and sarcomatoid renal cell carcinoma: CT and angiographic features. Radiology 1987;162:353?7. [ ] 13. Ro JY, Ayala AG, Sella A, Samuels ML, Swanson DA. Sarcomatoid renal cell carcinoma: clinicopathologic. A study of 42 cases. Cancer 1987;59: 516?26. [ ] 14. Cangiano T, Liao J, Naitoh J, Dorey F, Figlin R, Belldegrun A. Sarcomatoid renal cell carcinoma: biologic behavior, prognosis, and response to combined surgical resection and immunotherapy. J Clin Oncol 1999;15:523?8. [ ] 15. Bertoni F, Ferri C, Benati A, Bacchini P, Corrado F. Sarcomatoid carcinoma of the kidney. J Urol 1987;137:25?8. [ ] 16. Macke RA, Hussain MB, Imray TJ, Wilson RB, Cohen SM. Osteogenic and sarcomatoid differentiation of a renal cell carcinoma. Cancer 1985;56: 2452?7. [ ] 17. Nasu M, Yamazaki S, Kishi H, et al. Sarcomatoid clear renal cell carcinoma with condroid and osteoid differentiation: immunohistochemical and ultrastructural findings. J Urol Pathol 1997;6:171?80. [ ] 18. Bastacky S, McBec A, Fusca F, Becich MJ. Sarcomatoid renal cell carcinoma with malignant osseous and condroid differentiation: case report and literature review. J Urol Pathol 1996;5:119?38. [ ] 19. Levine E. Malignant renal parenchymal tumors in adults. In: Pollack HM, ed. Clinical urography ? an atlas and textbook of urological imaging. Philadelphia: WB Saunders, 1990:1216?91. [ ] 20. Prando D. Tumores malignos do parênquima renal. In: Prando A, Prando D, Caserta NMG, Bauab Jr T. Urologia ? diagnóstico por imagem. São Paulo: Sarvier, 1997:152?69. [ ]
Recebido para publicação em 25/11/2002
* Trabalho realizado no Departamento de Diagnóstico por Imagem e Anatomia Patológica do Hospital das Clínicas da Faculdade de Medicina da Universidade Federal de Goiás (HC-FMUFG), Goiânia, GO. |
|

Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554