Radiologia Brasileira - Publicação Científica Oficial do Colégio Brasileiro de Radiologia
AMB - Associação Médica Brasileira CNA - Comissão Nacional de Acreditação
 Vol. 38 nº 1 - Jan. / Fev. of 2005
Vol. 38 nº 1 - Jan. / Fev. of 2005
|
RELATO DE CASO
|
|
|
|
|
Autho(rs): Cyrillo Rodrigues de Araújo Júnior, Tarcísio Nunes Carvalho, Marlos Augusto Bitencourt Costa, Leonardo Valadares Lobo, Cristiano Rezio Fonseca, Kim-Ir-Sen Santos Teixeira |
|
|
Descritores: Fibrodisplasia ossificante progressiva, Miosite ossificante progressiva, Fisiopatogenia, Ossificação, Raios-X |
|
|
Resumo:
INTRODUÇÃO A fibrodisplasia ossificante progressiva (FOP) é uma doença rara, autossômica dominante, com expressão variável, que afeta todos os grupos étnicos(1-3). Sua prevalência é de 0,61 caso por um milhão de habitantes(4). Como na maioria dos casos não se observam antecedentes familiares, imagina-se ser esta doença devida a uma mutação esporádica(5). É uma enfermidade incapacitante em crianças e adultos jovens, basicamente caracterizada por osteogênese heterotópica progressiva e alteração congênita dos primeiros pododáctilos(6). A expressão fibrodisplasia ossificante progressiva substitui a antigo nome miosite ossificante(6), por ser a osteogênese ectópica ocorrendo no tecido conjuntivo, podendo afetar músculos, fáscias, ligamentos, tendões e cápsulas articulares. O doente geralmente apresenta ossificação progressiva do tecido conjuntivo, que causa uma limitação crescente da mobilidade osteoarticular, afetando principalmente a coluna vertebral, ombros, quadril e articulações periféricas. O paciente assume uma postura única e limitada, não podendo sequer sentar. Este aspecto caracteriza a forma avançada da doença, recebendo a denominação de síndrome do "stone man", antes descrita em apenas cerca de 600 doentes(7). Nesse estágio, o paciente rapidamente evolui para a morte, em conseqüência de problemas respiratórios de origem restritiva. Apresentamos um caso de FOP com 14 anos de evolução em um paciente de 22 anos de idade, do sexo masculino, manifestando grave comprometimento funcional, e discutimos os aspectos radiográficos característicos desta entidade.
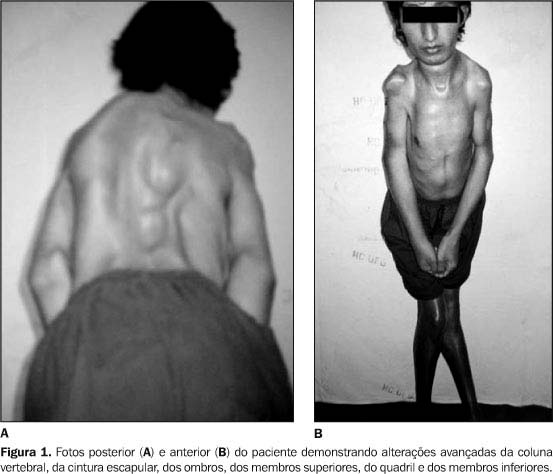
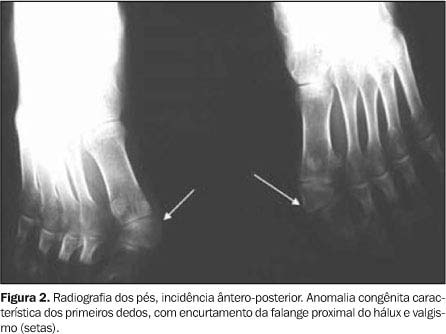
RELATO DO CASO Paciente de 22 anos de idade, branco, primeiro filho de uma prole de três (irmãos normais), sexo masculino, foi atendido em 21 de janeiro de 2002 no ambulatório de Ortopedia do Hospital das Clínicas da Faculdade de Medicina da Universidade Federal de Goiás (HC-FMUFG), referindo dor nas articulações e dificuldade para andar e principalmente ao sentar. Foi aparentemente saudável até os oito anos de idade, quando passou a ter episódios recorrentes de dor, rigidez das pernas e da coluna, evoluindo posteriormente para todo o corpo. O paciente não tinha antecedente pessoal ou familiar relevantes. No exame físico específico destacava-se a grande limitação funcional global do paciente, com rigidez universalmente distribuída, ausência de mobilidade do esqueleto axial e redução muito significativa da mobilidade das articulações periféricas. A articulação temporomandibular estava limitada e condicionava redução da abertura bucal. A coluna vertebral apresentava-se anquilosada, com escoliose torácica à esquerda, acentuação da lordose lombar e focos de ossificações ectópicas na musculatura paravertebral (Figura 1A). Havia redução da expansibilidade torácica, evidente rigidez nos quadris e ombros. Notavam-se importante atrofia muscular, adução das coxas, genuvalgo bilateral e pés em equinovaro (Figura 1B). Os primeiros dedos de ambos os pés eram significativamente mais curtos que os restantes e em valgismo (Figura 2).
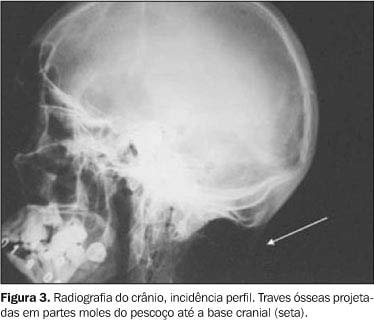
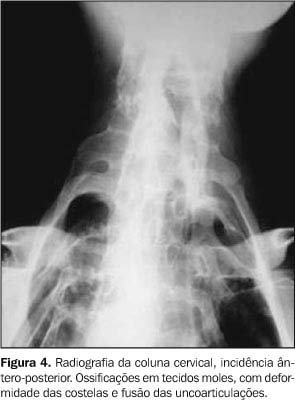
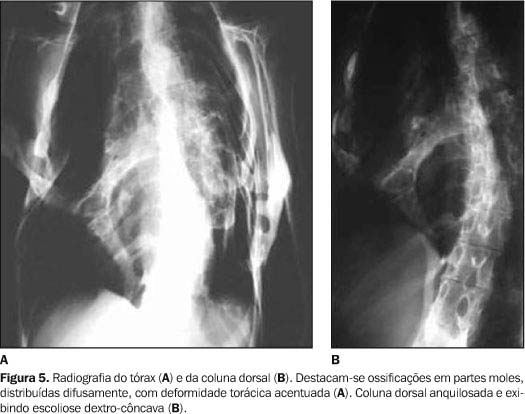
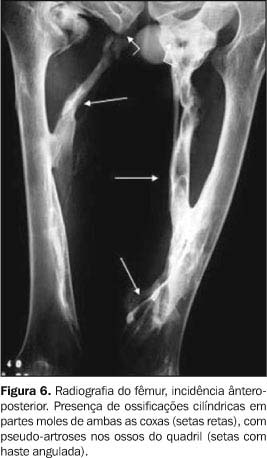
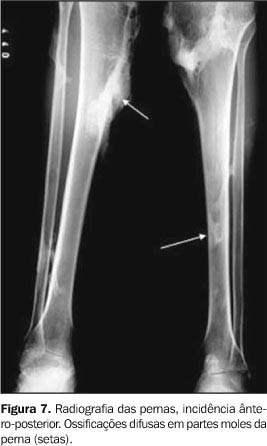
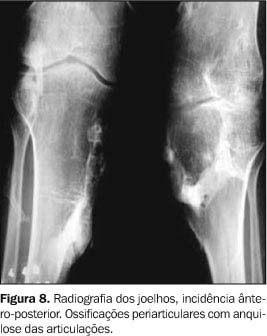
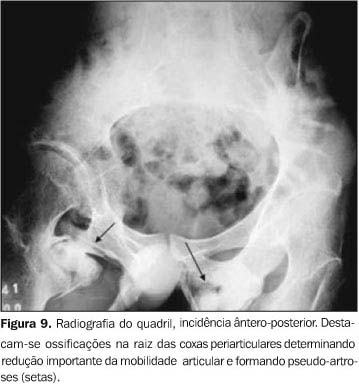
Os exames laboratoriais não mostraram alterações. As radiografias documentaram múltiplas calcificações em partes moles no pescoço (Figura 3), nos músculos paraespinhais (Figura 4 e 5), nos músculos adutores da coxa (Figura 6), difusamente em diversos grupos musculares das pernas (Figura 7) e periarticulares ao nível dos joelhos (Figura 8). Havia artrose das articulações do quadril, sendo identificadas, também, pseudo-artroses (Figuras 6 e 9), escoliose torácica à esquerda com anquilose (Figura 5) e encurtamento da falange proximal do hálux (Figura 2).
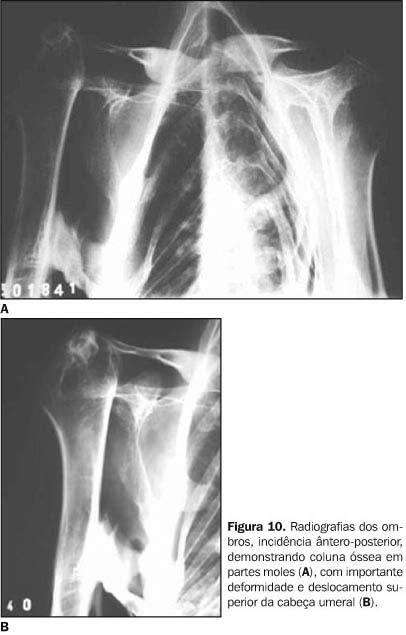
DISCUSSÃO Os sintomas da FOP são variáveis, com a maioria dos doentes apresentando calcificação das partes moles antes mesmo dos dez anos de idade. A idade média de início é em torno dos 3,6 anos(8). As primeiras manifestações localizam-se na coluna vertebral e nas articulações proximais. O quadro clínico caracteriza-se por sinais inflamatórios, por vezes acompanhados de expansões dolorosas, endurecimento dos tecidos periarticulares e perda progressiva da capacidade funcional da área afetada, sendo sua progressão no sentido axial-caudal e proximal-distal(9). Destaca-se o início quase sempre na musculatura espinhal superior(10). A evolução da doença pode conduzir à fusão das articulações costocondrais e o envolvimento assimétrico da musculatura paravertebral pode determinar o aparecimento de escoliose e doença pulmonar restritiva, uma das causas de freqüente morbidade e mortalidade desta doença(11). As radiografias convencionais documentam freqüentemente as anomalias esqueléticas constitucionais e as ossificações e anquiloses articulares correspondentes à história natural da doença. O primeiro achado radiográfico é a presença de expansões ou massas em tecidos moles, que gradualmente diminuem de tamanho e ossificam. A mineralização ocorre após três a quatro semanas, dando o aspecto final de colunas de osso que substituem os tecidos moles. Pseudo-artroses também podem ocorrer com essas colunas de osso, principalmente nos ombros e no quadril (Figuras 6, 9 e 10).
A FOP deve ser prontamente identificada baseando-se apenas na história clínica, no exame físico e nos achados radiográficos(6). Não deve ser realizado procedimento invasivo para determinação diagnóstica, pois tal fato é invariavelmente seguido de ossificações na região. A deformidade mais característica desta doença e que sempre deverá suscitar a hipótese de FOP é o encurtamento bilateral com valgismo dos primeiros pododáctilos (relatado em 79% a 100% dos pacientes)(12) (Figura 2); contudo, deve-se lembrar da existência do hálux valgo congênito isolado, entidade rara e que não faz parte da síndrome da FOP. Más-formações da mão também podem estar associadas, sendo observado encurtamento do primeiro metacarpiano e braquimesofalangismo com clinodactilia do dedo mínimo(13), alterações não observadas no nosso paciente. Os episódios de reagudização da FOP podem ocorrer espontaneamente ou podem ser precipitados por procedimentos invasivos como injeções, biópsias, flebopunções ou bloqueios anestésicos, daí a importância do diagnóstico preciso e não invasivo. A histopatologia desta doença varia com o tempo de evolução das lesões e só existem alterações nas áreas anatômicas afetadas. Este comportamento focal e evolutivo justifica a normalidade da biópsia obtida no paciente, numa fase inicial e provavelmente numa área não afetada(14). Nas lesões precoces há, geralmente, infiltrado linfocitário, macrófagos e fibroblastos, evoluindo mais tarde para áreas de tecido conjuntivo com ossificação central, nas quais se distinguem osteoblastos, osteócitos e osteoclastos(14). Recentemente, foi descrita a expressão aumentada da proteína morfogenética do osso 4 (BMP 4) nos fibroblastos presentes nas lesões precoces de FOP. A proteína BMP 4 está localizada no cromossomo 14q22-q23, onde estão sendo pesquisadas evidências da existência de mutações neste gene ou na região de seu promotor(15,16). Até o momento, não há tratamento conhecido e efetivo para esta doença. Toda conduta é conservadora e baseada no princípio do primum non nocere, evitando toda e qualquer condição potencialmente causadora de ossificação ectópica. Existem diversos estudos envolvendo quelantes do cálcio, extratos de hormônio paratireóideo e vitamina D, que não parecem ser promissores(14). Os corticóides poderão ser usados nos episódios de agudização inflamatória, embora não seja provada a inibição da ossificação ectópica. Etiodronato e isotretinoína também podem ser usados como inibidores da ossificação ectópica(14). O diagnóstico diferencial desta entidade é bem limitado, uma vez que seu fenótipo, a história clínica e os achados radiográficos praticamente fecham o quadro. Outras causas de ossificação ectópica devem sempre ser lembradas, tais como: osteodistrofia hereditária de Albright, calcificação heterotópica pseudomaligna, heteroplasia óssea progressiva e até mesmo o osteossarcoma(17,18).
CONCLUSÃO Apresentamos um caso de FOP com manifestações clínico-radiológicas muito características e com longo tempo de evolução, causando complicações tardias motoras e pulmonares usualmente descritas. Esta entidade é rara, existindo escassas referências na literatura brasileira. A importância do reconhecimento radiográfico desta enfermidade e de seus possíveis diagnósticos diferenciais foi abordada. Este caso ilustra bem a forma avançada da doença, com manifestações radiográficas características, e as dificuldades terapêuticas para estes pacientes. Foram discutidos, aqui, os últimos relatos da literatura ocidental sobre a fisiopatogenia e a genética desta incapacitante doença, que poderão eventualmente favorecer avanços na sua detecção precoce e terapêutica.
REFERÊNCIAS 1. Vastine JH, Vastine MF, Arango O. Myositis ossificans progressiva in homozigotic twins. AJR 1948;59:204-12. [ ] 2. Connor JM, Skirton H, Lunt PW. A three generation family with fibrodysplasia ossificans progressiva. J Med Genet 1993;30:687-9. [ ] 3. Delatycki M, Rogers JG. The genetics of fibrodysplasia ossificans progressiva. Clin Orthop 1998; 346:15-8. [ ] 4. Connor JM, Evans DA. Genetic aspects of fibrodysplasia ossificans progressiva. J Med Genet 1982;19: 35-9. [ ] 5. Becker PE, Von Knorre GV. Myositis ossificans progressiva. Ergeb Inn Med Kinderheilkd 1968;27: 1-8. [ ] 6. Buyse G, Silberstein J, Goemans N, Casaer P. Fibrodysplasia ossificans progressiva: still turning into wood after 300 years? Eur J Pediatr 1995;154: 694-9. [ ] 7. Nucci A, Queiroz LD, Santos AD, Camargo EE, Moura-Ribeiro MV. Fibrodysplasia ossificans progressiva. Arq Neuropsiquiatr 2000;58(2-A):342-7. [ ] 8. Rogers JG, Geho WB. Fibrodysplasia ossificans progressiva. A survey of forty-two cases. J Bone Joint Surg Am 1979;61:909-14. [ ] 9. Bridges AJ, Hsu K-C, Singh A, Churchill R, Miles J. Fibrodysplasia (myositis) ossificans progressiva. Semin Arthritis Rheum 1994;24:155-64. [ ] 10. Kaplan FS, McCluskey W, Hahn G, Tabas JA, Muenke M, Zasloff MA. Genetic transmission of fibrodysplasia ossificans progressiva. Report of a family. J Bone Joint Surg Am 1993;75:1214-20. [ ] 11. Buhain WJ, Rammohan G, Berger HW. Pulmonary function in myositis ossificans progressiva. Am Rev Respir Dis 1974;110:333-7. [ ] 12. Connor JM, Evans DA. Fibrodysplasia ossificans progressiva. The clinical features and natural history of 34 patients. J Bone Joint Surg Br 1982;64: 76-83. [ ] 13. Schroeder HW, Zasloff MA. The hand and foot malformations in fibrodysplasia ossificans progressiva. John Hopkins Med J 1980;147:73-8. [ ] 14. Fonseca JE, Evangelista T, Barroso L, Reis J, Gomes AR. Miosite ossificante progressiva: stone man. Acta Méd Port 2001;14:429-33. [ ] 15. Cramer SF, Ruehl A, Mandel MA. Fibrodysplasia ossificans progressiva: a distinctive bone-forming lesion of the soft tissue. Cancer 1981;48:1016-21. [ ] 16. Shafritz AB, Shore EM, Gannon FH, et al. Overexpression of an osteogenic morphogen in fibrodysplasia ossificans progressiva. N Engl J Med 1996;335:555-61. [ ] 17. Kaplan FS, Craver R, MacEwen GD, et al. Progressive osseous heteroplasia: a distinct developmental disorder of heterotopic ossification. Ttwo new case reports and follow-up of three previously reported cases. J Bone Joint Surg Am 1994;76:425-36. [ ] 18. Miller ES, Esterly NB, Fairley JA. Progressive osseous heteroplasia. Arch Dermatol 1996;132:787-91. [ ]
Recebido para publicação em 20/3/2003. Aceito, após revisão, em 22/1/2004.
* Trabalho realizado no Serviço de Diagnóstico por Imagem do Hospital das Clínicas da Faculdade de Medicina da Universidade Federal de Goiás (HC-FMUFG), Goiânia, GO. |
|

Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554