Radiologia Brasileira - Publicação Científica Oficial do Colégio Brasileiro de Radiologia
AMB - Associação Médica Brasileira CNA - Comissão Nacional de Acreditação
 Vol. 39 nº 6 - Nov. / Dez. of 2006
Vol. 39 nº 6 - Nov. / Dez. of 2006
|
RELATO DE CASO
|
|
|
|
|
Autho(rs): Anna Caroline Nobre Gomes, Cláudio Régis Sampaio Silveira, Roberto Guido Santos Paiva, Antônio Gilson Monte Aragão Jr., José Roberto Cavalcante Castro Jr. |
|
|
Descritores: Condrossarcoma, Osteocondromatose múltipla, Tomografia computadorizada, Fêmur |
|
|
Resumo:
INTRODUÇÃO A osteocondromatose múltipla, também chamada exostose hereditária múltipla ou aclasia diafisária, é um distúrbio da metáfise óssea, transmitido de modo autossômico dominante, que é geneticamente heterogêneo e que possui penetrância incompleta em indivíduos do sexo feminino(1). Schmale et al.(1) e Kivioja et al.(2) relatam prevalência do distúrbio de 1/50.000, embora outro estudo tenha mostrado prevalência de 9/1.000.000 na Europa(3). Os osteocondromas desenvolvem-se apenas nos ossos de origem endocondral e são o resultado da displasia periférica da placa de crescimento(1), causando deslocamento de áreas da placa. Esse é o tipo mais comum de tumor ósseo benigno(1) e acomete, principalmente, os ossos longos(1,4,5), pelve, escápula(2,4), tornozelo e joelho(1). A complicação mais séria apresentada pelos pacientes portadores de osteocondromatose múltipla é a degeneração maligna das exostoses cartilaginosas para condrossarcomas ou, mais raramente, para outros tipos de sarcomas. Há ampla variação da taxa de degeneração maligna descrita na literatura, sendo descrita como menor que 5% em estudo mais recente(6). Condrossarcomas periféricos são menos agressivos (ou possivelmente mais acessíveis à extirpação cirúrgica) que tumores centrais(5). O tratamento adequado implica ressecção completa do tumor e segmento do osso onde este surgiu. O prognóstico de pacientes com condrossarcoma desdiferenciado é pobre, independentemente da forma da terapia empregada; a maioria dos pacientes morre em conseqüência de metástases a distância por volta de um ano após o diagnóstico inicial(7). Neste trabalho, apresentamos um caso de condrossarcoma secundário grau I em uma paciente com osteocondromatose múltipla e revisão da literatura.
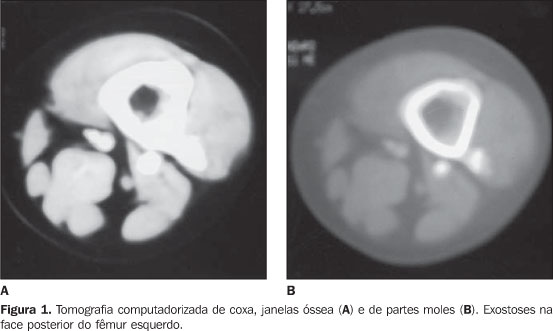
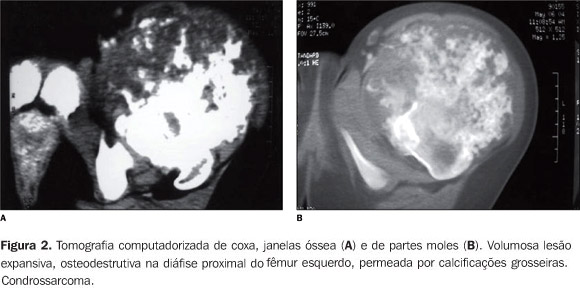
RELATO DO CASO Paciente do sexo feminino, 14 anos de idade, encaminhada ao ambulatório de ortopedia, com queixa de tumor na coxa esquerda. Paciente portadora de osteocondromatose múltipla, com tumoração na face lateral do terço proximal da coxa esquerda de crescimento lento e doloroso há dois anos. Não havia história familiar de exostoses múltiplas. Ao exame físico a paciente apresentava tumoração ocupando a face ântero-lateral do terço superior da coxa esquerda. A coxa esquerda apresentava diâmetro de 56 cm, e a direita, de 46 cm. Foi realizada, no nosso serviço, revisão da lâmina da biópsia feita no serviço de origem da paciente. Esta revisão confirmou o diagnóstico de condrossarcoma grau I em lesão condromatosa. Realizamos, em nosso serviço, exame de tomografia computadorizada (TC) com cortes axiais de 10 mm de espessura após administração de contraste iodado, que evidenciou lesão expansiva osteodestrutiva, acometendo a diáfise proximal, permeada por calcificações grosseiras e com componente sólido extravasando para partes moles, envolvendo o compartimento muscular anterior ao nível da raiz da coxa. A lesão deslocava o feixe músculo-nervoso femoral medialmente, mas não comprometia a perviedade vascular. A lesão media 13 × 12 × 11,5 cm nos planos ortogonais. Foram observadas excrescências ósseas compatíveis com osteocondromas na diáfise distal femoral lateral e medial, com ausência de rotura cortical. Devido ao extenso comprometimento de grupos musculares, de tecido celular subcutâneo e de pele, foi realizada desarticulação do colo femoral.
DISCUSSÃO Em 1786, John Huter fez a diferenciação entre a osteocondromatose solitária e a múltipla, sendo, portanto, o diagnóstico desta doença bem estabelecido atualmente(3). Em aproximadamente 10% dos casos documentados das séries de Schmale et al.(1) não havia história familiar de exostoses múltiplas, confirmando a informação já existente na literatura(4). Assim, o surgimento da doença foi implicado a uma mutação espontânea(4). No estudo de Schmale et al.(1) não foi detectada penetrância incompleta do distúrbio em indivíduos do sexo feminino, mas uma ausência de detecção de pequenas exostoses nesse grupo pela grande deposição de tecido gorduroso durante a puberdade. Os dados obtidos também sustentam o conceito de que o gene se expressa mais severamente em homens.
O achado radiológico diagnóstico de aclasia diafisária é a continuidade direta da massa com a cavidade medular do osso primário, com ausência de córtex subjacente. Isto se deve ao deslocamento da metáfise óssea durante seu crescimento através de um defeito no pericôndrio, com posterior crescimento de osso esponjoso dentro da massa à medida que os vasos invadem a cartilagem. O crescimento das exostoses ocorre durante a infância, cessando quando termina o crescimento da placa adjacente e podendo causar sintomas como resultado da compressão local de tecidos, e deformidades e alterações do comprimento ósseo(2). O crescimento de uma ou mais exostoses após o fim do período de crescimento esquelético deve alertar para a possibilidade de degeneração maligna do tumor benigno(5,6,8). Um sintoma muito comum é a dor(5,6,8), embora o crescimento tenha sido descrito como constante e indolor por Kivioja et al.(2), e a degeneração maligna, até como assintomática por Bovée et al.(6). Outros achados são complicações neurovasculares(8), fraturas patológicas(8) e espessamento acima de 1 cm da cápsula cartilaginosa em adultos(4,6,8). Devido ao risco que esses pacientes têm de desenvolver um tumor maligno, associado à dificuldade de detecção de sintomas de alarme em regiões como a pelve(2), aconselha-se que seja feito acompanhamento periódico desses pacientes com radiografias bianuais(2), principalmente daqueles com exostoses nos sítios mais comuns de degeneração sarcomatosa, ou seja, na pelve(1,2,4,5,8) e no ombro(1,5,8). Quando houver protuberâncias suspeitas, deve-se realizar uma ressonância magnética (RM)(2). Raramente os condrossarcomas acometem as partes distais dos membros. Essa complicação, portanto, costuma ocorrer após a puberdade, sendo rara durante a infância. Há uma maior freqüência de malignização entre 20 e 40 anos de idade(8), sendo o aumento do risco diretamente proporcional à idade(1). Os condrossarcomas podem ser primários ou secundários, estes surgindo como uma transformação maligna de um encondroma, ou, raramente, da cobertura cartilaginosa de um osteocondroma. Segundo o local, são subclassificados como intramedular e justacortical, e, histologicamente, como variantes convencional (hialina e/ou mixóide), de células claras, desdiferenciado e mesenquimal. Estes sarcomas geralmente têm história natural indolente, apresentando-se como dor e tumefação. O processo de transformação maligna ocorre numa freqüência de, aproximadamente, 5%(1,5). No entanto, a literatura mostra amplas variações dessa taxa. Taxas tão baixas quanto 0,6%(2) e tão elevadas quanto 25%(7) são também descritas, mas devem-se, provavelmente: a um viés de verificação e a uma incompleta detecção de indivíduos afetados que não desenvolveram sarcoma, mas que eram membros de família com múltiplas exostoses hereditárias(1); a um período de seguimento prolongado, o que aumenta o risco de degeneração sarcomatosa(1); a diferenças de idade entre as séries de estudo(5); ao critério de malignidade(5); e ao grau de especialização dos centros de onde vêm os casos(5). Radiografias, TC, RM, arteriografia e cintilografia óssea fornecem achados que sugerem o diagnóstico de condrossarcoma(8). O padrão de crescimento nodular da cartilagem produz recortes endosteais proeminentes radiograficamente. A matriz calcificada aparece como focos de densidade floculenta, observando-se calcificação mosqueada, em pipoca, pontilhada ou anular da matriz cartilaginosa(8). Radiografias seriadas geralmente dão alguma indicação de transformação maligna(4,5). Raramente, a radiografia é, como único exame, conclusiva; mais freqüentemente, deve-se ponderar a avaliação de todas as outras evidências(5). As margens bem definidas de uma lesão podem se tornar indistintas(4,8), ou a lesão pode aumentar de tamanho(4,5,8), ou a sua mineralização pode assumir o aspecto de vidro fosco(4). Relativa lucência dentro de uma região previamente mineralizada da capa cartilaginosa também implica a possibilidade de degeneração sarcomatosa(4,8). Embora a espessura da capa cartilaginosa desmineralizada seja, geralmente, menor que 1 cm em osteocondromas benignos, ela é, geralmente, maior que 2 cm quando ocorre transformação maligna(4). A evidência da espessura da capa cartilaginosa é, geralmente, mas nem sempre, um sinal confiável de malignidade ou de benignidade(4). Pode ser difícil avaliar essa espessura por meio de TC, devido à radiodensidade semelhante do pericôndrio, da bursa e de outros tecidos justapostos(4). Quanto mais radiotransparente o tumor, mais alta a probabilidade de que seja de alto grau. Um tumor de crescimento lento e de baixo grau causa espessamento reativo do córtex, enquanto uma neoplasia de alto grau mais agressiva destrói o córtex e forma uma massa de tecidos moles. A TC não é útil para diferenciar lesões benignas de malignas, embora uma TC negativa possa afastar a possibilidade de transformação maligna de uma exostose(2). Kivioja et al.(2) utilizaram, no seu estudo, a RM como técnica de screening para pelve, para fêmur proximal e para úmero superior, mas a sua utilização para detecção precoce de condrossarcoma e para seguimento dos pacientes com exostoses múltiplas é ainda muito limitada. Para Solomon(5), o problema do diagnóstico é agravado pela tentativa de se estabelecer uma precisa divisão entre a lesão benigna e o condrossarcoma. Por definição, pode-se aplicar o termo sarcoma somente para a neoplasia que tem a capacidade de metastatizar, e isso é o que se espera evitar com o tratamento adequado do tumor antes que ele manifeste essa característica(5). O que é importante, portanto, é avaliar o provável comportamento do tumor em vez de sua exata designação(5).
REFERÊNCIAS 1. Schmale GA, Conrad EU 3rd, Raskind WH. The natural history of hereditary multiple exostoses. J Bone Joint Surg Am 1994;76:986–992. [ ] 2. Kivioja A, Hervasti H, Kinnunen J, Kaitila I, Wolf M, Böhling T. Chondrosarcoma in a family with multiple hereditary exostoses. J Bone Joint Surg Br 2000;82:261–266. [ ] 3. Taniguchi K. A practical classification system for multiple cartilaginous exostosis in children. J Pediatr Orthop 1995;15:585–591. [ ] 4. Ostlere SJ, Gold RH, Mirra JM, Perlman RD. Case report 658: Chondrosarcoma of the proximal phalanx of right fourth finger secondary to multiple hereditary exostoses (MHE). Skeletal Radiol 1991;20:145–148. [ ] 5. Solomon L. Chondrosarcoma in hereditary multiple exostosis. South Afr Med J 1974;48:671–676. [ ] 6. Bovée JVMG, Sakkers RJB, Geirnaerdt MJA, Taminiau AHM, Hogendoorn PCW. Intermediate grade osteosarcoma and chondrosarcoma arising in an osteochondroma. A case report of a patient with hereditary multiple exostoses. J Clin Pathol 2002;55:226–229. [ ] 7. Kilpatrick SE, Pike EJ, Ward WG, Pope TL. Dedifferentiated chondrosarcoma in patients with multiple osteochondromatosis: report of a case and review of the literature. Skeletal Radiol 1997;26:370–374. [ ] 8. Merchan ECR, Sanchez-Herrera S, Gonzalez JM. Secondary chondrosarcoma. Four cases and review of the literature. Acta Orthop Belg 1993;59: 76–80. [ ]
Recebido para publicação em 3/3/2005.
* Trabalho realizado no Serviço de Radiologia do Hospital do Câncer do Ceará, Fortaleza, CE. |
|

Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554