Radiologia Brasileira - Publicação Científica Oficial do Colégio Brasileiro de Radiologia
AMB - Associação Médica Brasileira CNA - Comissão Nacional de Acreditação
 Vol. 39 nº 3 - Maio / Jun. of 2006
Vol. 39 nº 3 - Maio / Jun. of 2006
|
ARTIGO ORIGINAL
|
|
|
|
|
Autho(rs): Marcel Koenigkam Santos, Fernando Bortolato Faria, Clovis Simão Trad |
|
|
Descritores: Tomografia computadorizada, Doença intersticial pulmonar, Esclerose sistêmica |
|
|
Resumo:
INTRODUÇÃO A esclerose sistêmica é uma doença do tecido conjuntivo, crônica, multissistêmica, de etiologia não definida, caracterizada por fibrose, alterações degenerativas e anormalidades vasculares na pele (esclerodermia), articulações e órgãos viscerais, incluindo o trato gastrintestinal, pulmões, coração e rins. A característica proeminente na patogenia da esclerose sistêmica é a excessiva produção e o acúmulo de colágeno, envolvendo mecanismos imunológicos, lesão vascular e ativação de fibroblastos. É doença rara, sem preferência racial, com incidência estimada de 14,1 casos por milhão de habitantes por ano, mais freqüente no sexo feminino e infreqüente em crianças e homens jovens(1,2). A sobrevida é determinada pela intensidade da doença visceral e o acometimento pulmonar é a principal causa de morte. Dois terços dos pacientes com esclerose sistêmica apresentam doença pulmonar, com o sintoma respiratório mais comum sendo a dispnéia aos esforços, muitas vezes associada a tosse seca(1). Hipertensão arterial pulmonar está presente em 6% a 60% dos pacientes, mais comumente associada à doença pulmonar grave, porém pode acontecer isoladamente(3). Outras manifestações torácicas também podem ser encontradas, como espessamento e derrame pleural, dilatação esofágica e sinais de esclerose miocárdica. Classicamente, a doença pulmonar na esclerose sistêmica é descrita pelas evidências de fibrose acometendo as porções periféricas, posteriores e basais dos pulmões, com alterações inicialmente sutis que progressivamente aumentam e acometem os dois terços inferiores pulmonares, semelhante às apresentações da fibrose pulmonar idiopática e doença pulmonar da artrite reumatóide.
MATERIAIS E MÉTODOS Pacientes Foram retrospectivamente estudados os exames de imagem de 23 pacientes com diagnóstico de esclerose sistêmica, satisfazendo os critérios clínicos e anatomopatológicos, incluindo o fenômeno de Raynaud, biópsia de pele evidenciando a dermatoesclerose e o acometimento visceral, ainda excluindo-se outras doenças do colágeno como artrite reumatóide, lúpus eritematoso sistêmico e dermatomiosite/polimiosite. Todos os pacientes estudados apresentavam sintomas respiratórios, que levaram a equipe clínica a iniciar a investigação do acometimento pulmonar com exames de imagem e provas de função pulmonar. Dentre os 23 pacientes estudados, encontramos 21 mulheres, oito negros ou mulatos, com idade média de 47 anos ao início do comprometimento pulmonar. Um possível viés em nosso estudo pode ter ocorrido em razão do caráter terciário de nosso hospital, sendo os casos encaminhados ao serviço após avaliações em instituições primárias ou secundárias. Alguns pacientes encaminhados já possuíam queixas respiratórias crônicas, podendo, portanto, já apresentar doença pulmonar em estádio mais avançado. Com o objetivo de amenizar este problema, foram revistos os primeiros exames de imagem solicitados quando da suspeita de doença pulmonar. Exames de imagem Os exames de radiografia simples (RX) do tórax constavam de grafias com incidência póstero-anterior e perfil, feitas em aparelhos de RX convencionais. A tomografia computadorizada de alta resolução (TCAR) foi realizada em aparelho helicoidal (Somatom Emotion; Siemens), modo seqüencial, com cortes de 1,0 mm espaçados de 10,0 mm e reconstrução com filtro duro, com janelas para parênquima pulmonar e mediastino, sem a administração de contraste endovenoso, com o paciente em decúbito dorsal. A revisão dos exames foi realizada por pelo menos três médicos diferentes, incluindo médicos residentes e contratados do serviço de radiologia e diagnóstico por imagem do hospital. No RX observaram-se as características da opacidade pulmonar quando presente, incluindo o padrão (consolidação, reticulação, micronódulos) e a distribuição, além de sinais de perda de volume pulmonar. As alterações tomográficas foram classificadas em quatro padrões predominantes: opacidades em vidro fosco, opacidades reticulares, opacidades em vidro fosco associadas a opacidades reticulares e presença de faveolamento, observando-se a localização, a distribuição e a extensão do acometimento pulmonar. Outras alterações do parênquima pulmonar ou pleurais também foram avaliadas, assim como a presença de linfonodomegalia mediastinal e hilar, verificando-se a presença de nodos com menor diâmetro maior que 10 mm.
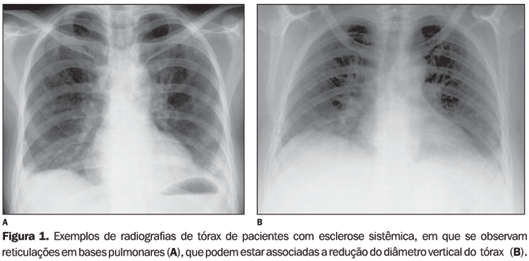
RESULTADOS No RX, o padrão reticular em bases pulmonares foi predominante, tendo sido verificado em 18 pacientes (78,2%). Em alguns casos, lesões mais extensas foram observadas, comprometendo porções superiores dos pulmões, além dos segmentos basais. Entre estes verificaram-se, também, casos com redução do diâmetro vertical do tórax, sinal de perda volumétrica por retração diafragmática, característica que marca as doenças pulmonares que cursam com fibrose nos lobos basais (Figura 1).
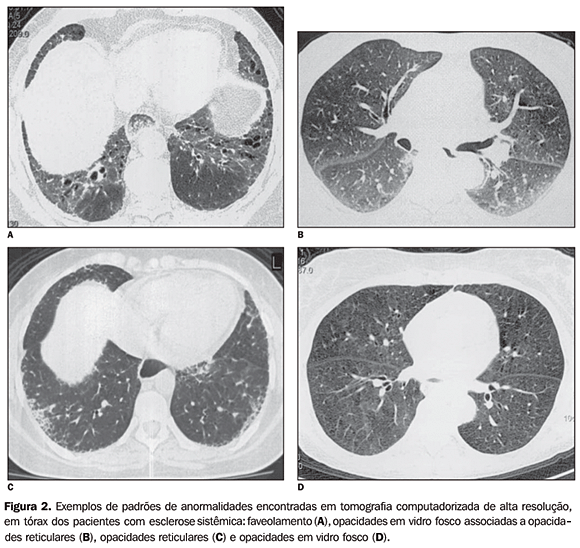
A TCAR evidenciou lesão pulmonar em todos os pacientes estudados, encontrando-se faveolamento em nove pacientes (39,1%), opacidades em vidro fosco associadas a opacidades reticulares em oito (34,7%), predomínio de opacidades reticulares em cinco (21,7%) e vidro fosco em um paciente (4,3%). As lesões verificadas na TCAR predominaram nos segmentos pulmonares basais, comprometendo principalmente o parênquima periférico e posteriormente (Figura 2). Outras alterações visualizadas em alguns casos foram bronquiectasias de tração, bolhas e focos de consolidação pulmonar. A presença de linfonodos mediastinais ou hilares com menor diâmetro que 10 mm foi verificada em cinco pacientes (21%).
DISCUSSÃO Evidência de doença pulmonar tem sido descrita em RX de tórax em 20% a 65% dos pacientes com esclerose sistêmica(2). Nosso estudo evidenciou alterações em 78,2% dos casos, desde alterações sutis como finas reticulações em bases pulmonares até lesões extensas com sinais de perda de volume pulmonar. Uma freqüência pouco maior de alterações radiográficas em nosso estudo pode ser justificada pelo caráter terciário de nosso hospital. A TCAR freqüentemente mostra comprometimento pulmonar nos pacientes com suspeita clínica e exames de RX normais, com alterações sutis ou duvidosas(4). O padrão de anormalidades tomográficas possui boa correlação com os achados histopatológicos, diferenciando padrões com predomínio de processo inflamatório (opacidades em vidro fosco) de lesões predominantemente fibróticas (opacidades reticulares e faveolamento), com os padrões inflamatórios estando associados a uma resposta superior ao tratamento(5). A TCAR mostra, ainda, correlação com os resultados de testes de função pulmonar, evidenciando doença pulmonar restritiva e distúrbio de difusão, através, principalmente, da redução dos valores do volume pulmonar e da capacidade de difusão para o monóxido de carbono, respectivamente(6-8). Não há estudos que correlacionam as alterações tomográficas e a idade do paciente ou o tempo de duração da doença. Nosso estudo evidenciou lesões pulmonares nos 23 pacientes estudados, com maior proporção de faveolamento, seguido de opacidades em vidro fosco associadas a opacidades reticulares, e uma menor proporção de predomínio de opacidades reticulares, enquanto o predomínio de opacidades em vidro fosco isoladamente foi incomum. Dessa maneira, observaram-se alterações sugestivas de fibrose na maior parte dos casos (faveolamento e opacidades reticulares somando 60,8%), porém com boa parte apresentando padrões sugestivos de processo inflamatório. Linfonodomegalia torácica está freqüentemente associada à doença pulmonar na esclerose sistêmica, porém mostra relação não com o padrão de anormalidades vistas na TCAR, mas sim com a extensão do comprometimento do parênquima pulmonar(9). Nosso estudo mostrou linfonodomegalia em cinco (21%) dos 23 casos, em quatro casos associada a extenso comprometimento pulmonar. Os estudos histopatológicos iniciais, baseados nas alterações observadas em cadáveres, descrevem semelhança quase indistinguível entre a esclerose sistêmica e a fibrose pulmonar idiopática(10), porém o prognóstico e a resposta terapêutica são melhores nos pacientes com acometimento pulmonar da esclerose sistêmica. Recentemente, as pneumonias intersticiais foram reclassificadas, organizadas em um grupo heterogêneo de doenças inflamatórias, baseado em seus padrões histopatológicos e com apresentações clínicas e radiológicas diferentes entre si(11,12). A apresentação clínica da fibrose pulmonar idiopática é descrita patologicamente como pneumonia intersticial usual, marcada pela fibrose intersticial grosseira, predominando no parênquima subpleural das regiões basais, correspondendo ao padrão de faveolamento visto na TCAR. A biópsia pulmonar nos casos de esclerose sistêmica não faz parte da rotina clínica, porém alguns estudos têm descrito a pneumonia intersticial não-específica como padrão histopatológico predominante, encontrando as alterações compatíveis com pneumonia intersticial usual, ou seja, fibrose pulmonar idiopática, em menor proporção dos casos(13,14). Como já citado anteriormente, existe boa correlação entre os achados tomográficos e as alterações histopatológicas das doenças pulmonares, inclusive quanto à diferenciação entre a pneumonia intersticial usual e a pneumonia intersticial não-específica, sendo a última caracterizada por predomínio de opacidades em vidro fosco e finas opacidades reticulares, diferentemente do faveolamento basal e periférico observado na primeira(15). Vários estudos recentes, incluindo este, têm demonstrado que as lesões pulmonares descritas em TCAR de pacientes com esclerose sistêmica são menos extensas e com uma maior proporção de alterações sugestivas de processo inflamatório do que as encontradas nos pacientes com fibrose pulmonar idiopática, ou seja, o quadro tomográfico da esclerose sistêmica é mais semelhante ao da pneumonia intersticial não-específica idiopática(16). Portanto, deve ser salientada a importância da TCAR nos pacientes com esclerose sistêmica, não somente como meio diagnóstico eficaz, detectando alterações precoces, mas também na avaliação prognóstica, identificando aqueles com alterações de predomínio de processo inflamatório, que apresentam melhor resposta a terapêutica.
REFERÊNCIAS 1.Gilliland BC. Systemic sclerosis (scleroderma). In: Braunwald E, Fauci AS, Kasper DL, Hauser SL, Longo DL, Jameson JL, editors. Harrison's Principles of internal medicine. 15th ed. New York: McGraw-Hill, 2001. [ ] 2.Arroliga AC, Podell DN, Matthay RA. Pulmonary manifestations of scleroderma. J Thorac Imaging 1992;7:30-45. [ ] 3.Ungerer RG, Tashkin DP, Furst D, et al. Prevalence and clinical correlates of pulmonary arterial hypertension in progressive systemic sclerosis. Am J Med 1983;75:65-74. [ ] 4.Warrick JH, Bhalla M, Schabel SI, Silver RM. High resolution computed tomography in early scleroderma lung disease. J Rheumatol 1991;18: 1520-1528. [ ] 5.Ooi GC, Mok MY, Tsang KWT, et al. Interstitial lung disease in systemic sclerosis: an HRCT-clinical correlative study. Acta Radiol 2003;44:258-264. [ ] 6.Remy-Jardin M, Remy J, Wallaert B, Bataille D, Hatron PY. Pulmonary involvement in progressive systemic sclerosis: sequential evaluation with CT, pulmonary function tests and bronchoalveolar lavage. Radiology 1993;188:499-506. [ ] 7.Kim EA, Johkoh T, Lee KS, et al. Interstitial pneumonia in progressive systemic sclerosis: serial high-resolution CT findings with functional correlation. J Comput Assist Tomogr 2001;25:757-763. [ ] 8.Shahin AA, Sabri YY, Mostafa HA, et al. Pulmonary function tests, high-resolution computerized tomography, alpha1-antitrypsin measurement, and early detection of pulmonary involvement in patients with systemic sclerosis. Rheumatol Int 2001;20:95-100. [ ] 9.Bhalla M, Silver RM, Shepard JA, McLoud TC. Chest CT in patients with scleroderma: prevalence of asymptomatic esophageal dilatation and mediastinal lymphadenopathy. AJR Am J Roentgenol 1993;161:269-272. [ ] 10.D'Angelo WA, Fries JF, Masi AT, Shulman LE. Pathologic observations in systemic sclerosis (scleroderma). A study of fifty-eight autopsy cases and fifty-eight matched controls. Am J Med 1969; 46:428-440. [ ] 11.Müller NL, Colby TV. Idiopathic interstitial pneumonias: high resolution CT and histologic findings. RadioGraphics 1997;17:1016-1022. [ ] 12.Katzenstein AA, Myers JL. State of the art: idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1998;157:1301-1315. [ ] 13.Bouros D, Wells AU, Nicholson AG, et al. Histopathologic subsets of fibrosing alveolitis in patients with systemic sclerosis and their relationship to outcome. Am J Respir Crit Care Med 2002;165:1578-1579. [ ] 14.Kim DS, Yoo B, Lee JS, et al. The major histopathologic pattern of pulmonary fibrosis in scleroderma is nonspecific interstitial pneumonia. Sarcoidosis Vasc Diffuse Lung Dis 2002;19:121-127. [ ] 15.MacDonald SL, Rubens MB, Hansell DM, et al. Nonspecific interstitial pneumonia and usual interstitial pneumonia: comparative appearances at and diagnostic accuracy of thin-section CT. Radiology 2001;221:583-584. [ ] 16.Desai SR, Veeraraghavan S, Hansell DM, et al. CT features of lung disease in patients with systemic sclerosis: comparison with idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia. Radiology 2004;232:560-567. [ ]
Recebido para publicação em 9/6/2005.
* Trabalho realizado no Centro de Ciências das Imagens e Física Médica do Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo, Ribeirão Preto, SP, Brasil. |
|

Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554