Radiologia Brasileira - Publicação Científica Oficial do Colégio Brasileiro de Radiologia
AMB - Associação Médica Brasileira CNA - Comissão Nacional de Acreditação
 Vol. 43 nº 3 - May / June of 2010
Vol. 43 nº 3 - May / June of 2010
|
WHICH IS YOUR DIAGNOSIS?
|
|
|
|
|
Autho(rs): Jaílson Lopes, Marcelo Bordalo-Rodrigues |
|
|
IMD, Resident, Division of Musculoskeletal System at Instituto de Radiologia do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (InRad/HC-FMUSP), São Paulo, SP, Brazil
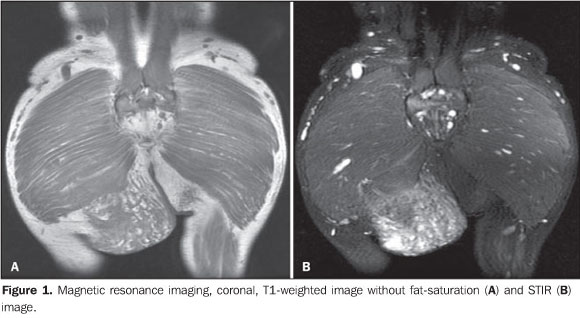
A male, 29-year old, black patient presented with a mass on his right gluteal region that had developed during his first infancy. The mass size was unchanged for at least ten years and the patient was asymptomatic and seeking clinical assistance for aesthetic reasons, being referred to the Service of Radiology at Instituto de Ortopedia e Traumatologia (IOT) of Faculdade de Medicina da Universidade de São Paulo by the IOT group of tumors to rule out the possibility of a malignant neoplastic process in the right gluteal region. Images description Figure 1. Magnetic resonance imaging, coronal, T1-weighted image without fat saturation (A) and STIR image (B) demonstrate an expansile heterogeneous, ill-defined mass in contact with the right gluteus maximus and the ischio-rectal fossa, infiltrating the subcutaneous tissue, com intermediate signal intensity on T1-weighted, and high signal intensity on STIR images.
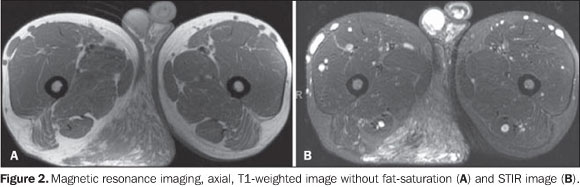
Figure 2. MRI, axial, T1-weighted image without fat-saturation (A) and STIR image (B) demonstrate an expansile mass in the ischio-rectal fossa in contact with perineal structures and infiltrating the subcutaneous tissue, with intermediate signal intensity on T1-weighted, and high signal intensity on STIR images.
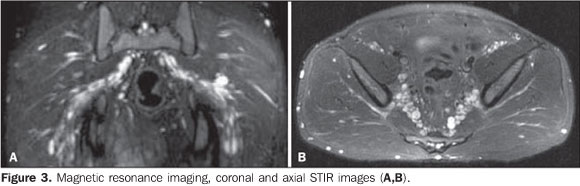
Figure 3. MRI, coronal and axial STIR images (A,B) demonstrate bilateral, symmetrical, well-defined, enchained fusiform structures adjacent to the bilateral piriform muscle, typical of deep plexiform neurofibromas.
Diagnosis: Plexiform neurofibroma - deep and superficial variances in a patient with type 1 neurofibromatosis.
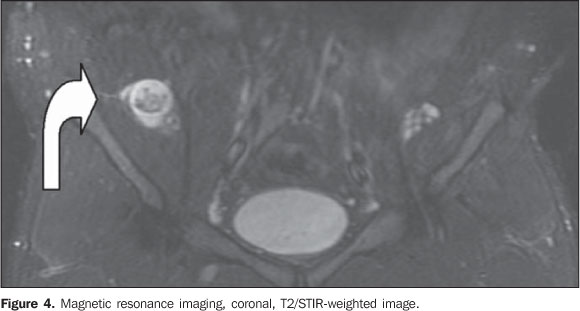
COMMENTS Type 1 neurofibromatosis (NF1) is an autosomal dominant disease that in more than 90% of cases is associated with a sporadic or inherited NF1 gene mutation, affecting one in every 3,500 individuals, representing the most common phakomatosis(1). This disease presents a range of focal and, most frequently, systemic signs in the chest, abdomen, pelvis and limbs, among others(1,2). The diagnosis is clinical, and generally the disease onset occurs in the childhood but in about 10% of cases a late onset is observed in a frustrating or atypical form of neurofibromatosis(3). Classically, neurogenic tumors are found out of the central nervous system and include the typical (non-plexiform) neurofibromas and plexiform neurofibromas (PNF). These tumors are benign and originate from the connective tissue of the nervous sheaths, particularly the endoneurium(2). The typical neurofibroma presents a hyperintense halo on T2-weighted sequences, with low signal intensity in the center, that may be less intense according to the amount of fibrous tissue within the mass, corresponding to the classical target sign(4), and that was also observed in the present case (Figure 4, curved arrow).
Among the clinical findings of NF1, the presence of a PNF itself is sufficient to complete the diagnosis. The term "plexiform" refers to a network-like pattern of growth, with the tumor development in multiple nerve fascicles, this nerve becoming thickened and surrounded by a proteinaceous matrix forming nodules or masses(4). In the spectrum of NF1, the frequency of PNFs may range from 25% to 30%(5,6), but other authors consider such statistics as underestimated as just clinical findings are taken into consideration(3,4). Frequently, PNFs involve nervous plexuses and dorsal roots, as well as other deep structures, particularly major nerves(7). Generally, PNFs are present at birth, becoming more noticeable in a later phase(5). Such disease may be asymptomatic but, differently from typical neurofibromas, cause aesthetic problems, pain and functional disorders, with a remarkable morbidity because of the continuous growth of the mass and about 10% o risk for transforming into a malignant nerve sheath tumor(8). PNFs are classified according to their location as follows: superficial PNFs, those arising between the skin and the muscle fascia; and deep PNFs, those arising underneath the muscle fascia. Some lesions may be located in both compartments and the site of their largest volume (> 50%) will define their categorization(7). Superficial and deep PNFs present different characteristics which can be clearly recognized on imaging studies such as magnetic resonance imaging (MRI), highlighting that, generally, superficial PNFs are unilateral, presenting as ill-defined, infiltrating masses, with a diffuse pattern and no target sign (Figures 1 and 2); and the deep ones tend to be bilateral, presenting as well-defined masses, with a nodular, target or fascicular pattern, also observed in the present case (Figure 3). Increased vascularization is observed in both types of PNF. High signal intensity is observed on T2/STIR-weighted sequences, but high/intermediate signal intensity may be observed on T1-weighted sequences because of the proteinaceous content of the tumor. Gadolinium presents a highly variable enhancement pattern, thus gadolinium enhanced imaging is not utilized as a standard protocol(7,9). These tumors do not require histopathological analysis, since clinical and imaging criteria are satisfactory to achieve a diagnosis. A tailored, individualized therapeutic management is required, focusing on the greatest possible aesthetic satisfaction, functional rehabilitation, pain and neurological deficit management. A fast growth of the mass and persistent pain draw the attention to a possible malignization(1,7,9).
CONCLUSION The imaging recognition superficial plexiform neurofibromas allows the establishment of a diagnosis of type 1 neurofibromatosis, in many situations ruling out the presence of a malignant process.
REFERENCES 1. Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol. 2009;61:1-14. [ ] 2. Fortman BJ, Kuszyk BS, Urban BA, et al. Neuro-fibromatosis type 1: a diagnostic mimicker at CT. Radiographics. 2001;21:601-12. [ ] 3. Jett K, Friedman JM. Clinical and genetic aspects of neurofibromatosis 1. Genet Med. 2010;12:1-11. [ ] 4. Korf BR. Plexiform neurofibromas. Am J Med Genet. 1999;89:31-7. [ ] 5. Huson SM, Harper PS, Compston DAS. Von Recklinghausen neurofibromatosis. A clinical and population study in south-east Wales. Brain. 1988;111(Pt 6):1355-81. [ ] 6. Tonsgard JH, Kwak SM, Short P, et al. CT imaging in adults with neurofibromatosis-1: frequent asymptomatic plexiform lesions. Neurology. 1988;50:1755-60. [ ] 7. Lim R, Jaramillo D, Poussaint TY, et al. Superficial neurofibroma: a lesion with unique MRI characteristics in patients with neurofibromatosis type 1. AJR Am J Roentgenol. 2005;184:962-8. [ ] 8. Rasmussen SA, Yang Q, Friedman JM. Mortality in neurofibromatosis 1: an analysis using U.S. death certificates. Am J Hum Genet. 2001;68: 1110-8. [ ] 9. Mautner VF, Hartmann M, Kluwe L, et al. MRI growth patterns of plexiform neurofibromas in patients with neurofibromatosis type 1. Neuro-radiology. 2006;48:160-5. [ ] Study developed at Instituto de Ortopedia e Traumatologia (IOT) and at Instituto de Radiologia (InRad) of Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (HC-FMUSP), São Paulo, SP, Brazil. |
|

Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554